







1. Introduction
Polyurethane (PU) elastomers, synthesized via the polyaddition reaction between polyisocyanates and macropolyols, have been a cornerstone in various industries. Their unique chemical structure offers unparalleled flexibility, durability, and resistance to chemicals and impact [1]. These properties have made PU indispensable in applications ranging from foam insulation to lightweight construction, coatings, and more [2]. However, the majority of traditional PUs are derived from petroleum resources, a non-renewable source. This reliance on petroleum not only poses challenges to sustainable development but also raises concerns about the depletion of these resources [3].
In the quest for sustainability, Bio-based Polyurethane (BPU) has emerged as a beacon of hope. Synthesized using renewable biomass such as plant oil, plant protein, and wood fibers, BPUs represent a paradigm shift from traditional petroleum-based PUs [4]. The environmental benefits of BPUs are manifold. Their production generates lower emissions, utilizes sustainable biomass feedstocks, and aligns with global environmental protection principles [5]. Moreover, BPUs offer health and safety benefits. Being derived from renewable biomass and devoid of toxic substances in their production, BPUs pose lower risks to human health and the environment [6].
Yet, the journey of BPUs from research labs to mainstream markets is fraught with challenges. A significant hurdle is their mechanical properties. BPUs often exhibit inferior mechanical properties compared to traditional PUs. Their molecular structure is relatively loose, has a wider molecular weight distribution, and exhibits lower crystallinity [7]. These structural nuances can adversely affect the material’s mechanical properties, limiting its application in sectors that demand robust materials, such as automotive components or building materials [8].
The recyclability and biodegradability of BPUs are other areas that need attention. A majority of BPUs are thermosetting plastics, which are challenging to dissolve and recover using organic solvents [9]. Although they are derived from biological resources, their structure often impedes effective degradation in natural environments [10]. This poses a significant challenge, especially when the world is grappling with plastic pollution and is in dire need of materials that are both high-performing and environmentally friendly [11].
To address the mechanical limitations of BPUs, researchers have explored various methods. These include the use of fillers, cross-linking agents, or blending with other polymers [12]. While these methods have shown promise in enhancing the mechanical properties of BPUs, they often introduce other challenges. For instance, the addition of fillers can compromise the recyclability of the elastomer, while excessive cross-linking can reduce the material’s overall flexibility [13]. Blending with other polymers can lead to phase separation issues, affecting the uniformity and consistency of the resulting material [14]. Calvo-Correas et al. synthesized PU using chain extenders derived from corn and sugar, combined with petroleum-based crosslinkers, to polymerize macrolipids and diisocyanates [10]. Despite the bio-based approach, the resultant PU lacked the anticipated biodegradability, undermining its environmental benefits. Concurrently, Zhao et al. utilized vanillin, obtained from lignin cleavage, as a precursor, modifying it to produce VBD. They then synthesized thermosetting BPU using stage II cyanate and trimethylpropane [2]. While these BPUs exhibited pronounced biodegradability, their recyclability remains contentious. The challenge lies in enhancing the mechanical properties of thermosetting BPUs without compromising recyclability. Historically, research prioritized elastomer strength and elongation over toughness, often overlooking the issue of low bio-content. For instance, Gang et al. synthesized BPU by chemically processing lignin and incorporating nicilin [15]. By introducing 5-20 mol% DV-EA, derived from vanillin dimer and ethanolamine, they enhanced the BPU’s mechanical properties through additional hydrogen bonding. However, the limited inclusion of DV-EA resulted in a persistently low bio-content, hindering the reduction of petroleum-based PU usage. In summary, achieving a balance between mechanical robustness, biodegradability, and recyclability in BPU remains an unresolved challenge in current research.
In light of these challenges, our research introduces a novel approach. Drawing inspiration from recent studies, we explore the potential of a carefully engineered A-B-A triblock polymer mixed with castor oil polyols as the BPU soft segment [16]. This approach aims to enhance the mechanical properties of BPUs, ensuring they are both recyclable and biodegradable. The A block is strategically designed to possess improved compatibility with the hard segment, allowing for efficient adjustment of the regularity of the hard segment. The B block exhibits enhanced crystallinity compared to other components. The presence of crystallinity in the B block of BPU enables it to serve as a physical cross-linking site, thereby enhancing the mechanical properties.
Herein, we employed a strategic approach to synthesize a BPU endowed with optimal characteristics. We utilized the PCL-PEG-PCL triblock polymer, where the PCL block, acting as the ‘A’ segment, modulates the hard segment’s size and imparts biodegradability to the BPU. Conversely, the PEG block, serving as the ‘B’ segment, functions as a physical cross-linking site, constraining the freedom of molecular chain segments. And the bio-based polyol was 1910-S which was obtained from ZheJiangHyaFon New Materials Co., Ltd.. The synthesis process is depicted in Figure 1(a). Initially, 1910s and tri-block polyols with 1,5-Pentane diisocyanate (PEDI) were combined to form a prepolymer. These prepolymers were subsequently blended in a 1:1 ratio. The synthesis was completed using 1,3-propylene glycol as a chain extender. To further investigate the impact of block B’s crystallinity on BPU performance, PTMEG and PDMS were chosen as comparative samples for the PEG block. Comprehensive evaluations, including tensile strength, biodegradation, and recyclability tests, were conducted on the synthesized BPU. The findings revealed that our BPU showcased remarkable toughness, recyclability, and biodegradability, positioning it as a viable contender in the realm of sustainable materials. The structure of our designed BPU is shown in Figure1(b).
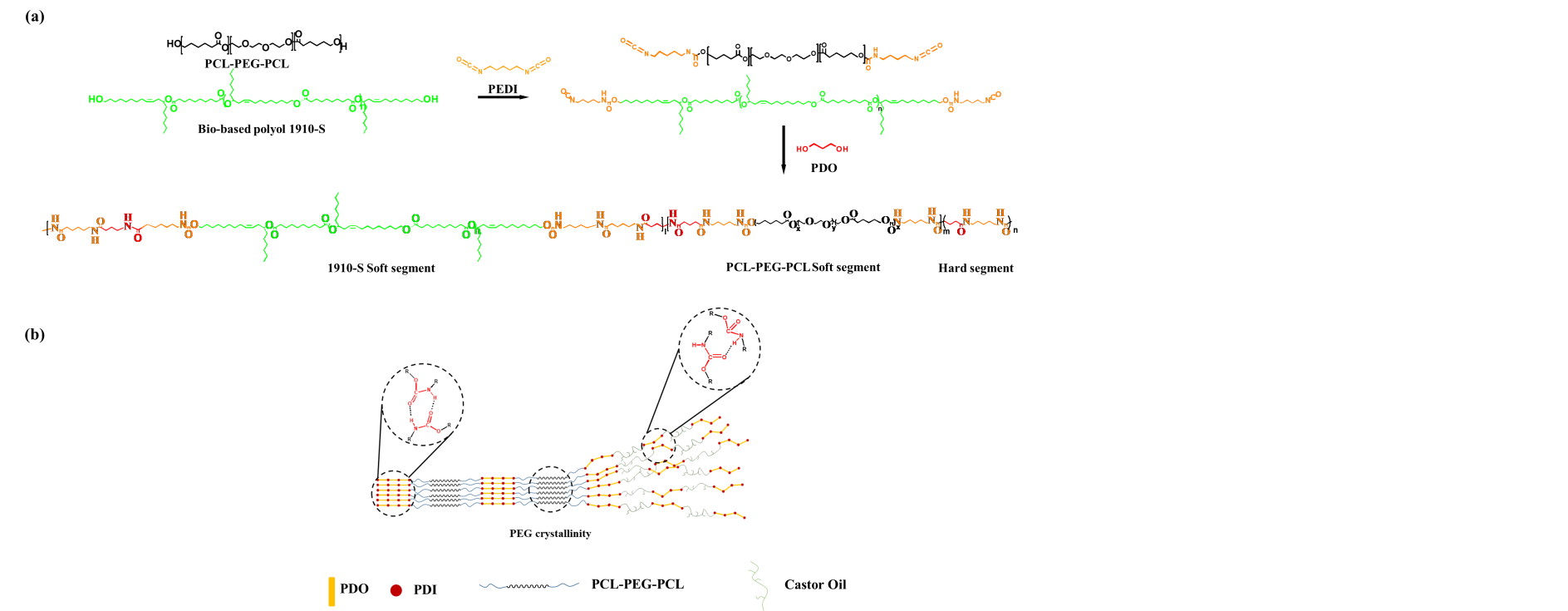
Figure 1. Overview of BPUPEG synthesis and structure. (a) Two-step synthesis routine of BPUPEG. (b) Schematic illustration of the structure of the triblock polymer-modified BPUPEG.
2. Materials and Experimental Section
2.1. Materials
Polyethylene glycols (OH-PEG-OH, Mn ≈ 1050, PDI= 1.09) was purchased from Alfa Aesar Co., Ltd. ε-Caprolactone (≥ 99.9%) was purchased from Ju Ren Co., Ltd. Poly(dimethyl siloxane) diols and poly(dimethyl siloxane) diols (HO-PDMS-OH, Mn ≈ 1020 and 1950, PDI = 1.25 and 1.21) were purchased from Tanguitech Co., 1,5-Pentane diisocyanate (PEDI, ≥ 99.8%) was purchased from BASF Co., Ltd. 1,3-Butane diol (PDO, ≥ 99.9%) was purchased from Aladdin Co., Ltd. Benzoyl Chloride (≥ 99.5%) was purchased from Adamas Co., Ltd. Bio-based soft segment 1910-S was synthetized via ZheJiangHyaFon New Materials Co., Ltd.
2.2. Methods
2.2.1. Synthesis of the tri-block co-polymer diols. HO-PCL-PTMEG-PCL-OH, HO-PCL-PEG-PCL-OH and HO-PCL-PDMS-PCL-OH were synthesized via ring-opening polymerization of ε-caprolactone using HO-PTMEG-OH (Mn ≈ 1050, PDI = 1.15), HO-PEG-OH (Mn ≈ 1050, PDI = 1.09) and HO-PDMS-OH (Mn ≈ 1020, PDI = 1.25) as the macromolecular initiators, respectively. Specifically, the macromolecular initiator (50 wt.%), ε-caprolactone (50 wt.%) and additional n-butyl titanate (TBT, 25 ppm) were fed into a glass reactor equipped with a vacuum pump and oil bath under nitrogen atmosphere at room temperature. With stirring, the system was allowed to react at 120 ℃ for 17 h, followed by application of vacuum under 150 mbar and 135 ℃ for 3 h. The final product was cooled down to 80 ℃, filtered, packaged and sampled for determination. The molecular structure and weight of HO-PCL-PEG-PCL-OH, HO-PCL-PTMEG-PCL-OH, HO-PCL-PDMS-PCL-OH and 1910-S are presented in Figure S1 and S2 to demonstrate successful synthesis.
2.2.2. Synthesis of BPU samples. BPU based on different A-B-A triblock polymer blended with castor oil as soft segments were prepared following a systematic approach, as illustrated in Scheme 1(b). Taking BPUPCL-PEG-PCL as an example, the detailed steps of BPU synthesis are introduced in detail. In this study, a two-step approach was employed to successfully synthesise BPU. To begin with, appropriate counterparts of PCL-PEG-PCL and castor oil polyols were subjected to a blocking reaction with PEDI, resulting in the formation of prepolymers. The experimental protocol entails conducting the reaction at a temperature of 85 °C for 6 hours, while ensuring an inert atmosphere of argon is maintained throughout the process. Subsequently, the prepolymer comprising of PCL-PEG-PCL and castor oil polyol was homogeneously blended in a precise mass ratio of 1:1. Subsequently, a certain amount of chain extender PDO was introduced into the blended prepolymer matrix. To achieve effective mixing and eliminate any potential air bubbles or dissolved gas, a Flacktec speedmixer operating at 2500 rpm was employed for 1 minute. Subsequently, the mixture was poured into the preheated molds and maintained at 110 ℃ for 14 hour, followed by controlled cooling to below 60 ℃. The resulting BPU samples were then meticulously demolded using gloves to prevent any damage. The BPU samples were named as BPUx, where x represents the B block of triblock copolymer. Specially, BPU1910-S represents the all the soft segments of BPU are castor oil polyols.
2.3. Characterization
2.3.1. Nuclear Magnetic Resonance Spectroscopy (NMR). 1H nuclear magnetic resonance spectra were obtained with a Bruker AV400 MHz NMR spectrometer at 298 K. Samples (3-5 mg) were dissolved in 0.55 mL of CDCl3, with tetramethyl silane (TMS) as the standard.
2.3.2. Gel Permeation Chromatography (GPC). GPC (Agilent/Wyatt 1260) was used to determine the molecular weights (Mw.) and PDI of the BPUs at 30 °C. Anhydrous N,N-Dimethylformamide (DMF) for HO-PEG-OH and HO-PCL-PEG-PCL-OH, anhydrous Tetrahydrofuran (THF) for HO-PCL-PDMS-PCL-OH was used as the elution phase at a flow rate of 1.0 mL/min, respectively. Narrow polyethylene oxide standards with molecular weights ranging from 550 to 100,000 g/mol were used to calibrate the system.
2.3.3. Attenuated Total internal Reflection Infrared Spectroscopy (ATR-IR). The BPUs were characterized by ATR-IR (Thermofisher Nicolet 6700) using a KRS-5 crystal with a 2.4 refractive index at a 45° angle. The resolution was 2.0 cm-1, and 64 scans were conducted.
2.3.4. Differential Scanning Calorimetry (DSC). The thermal attributes of the soft and hard segments, as well as the synthesized BPUs, were examined using DSC (Mettler Toledo 822e). Samples (~5-10 mg) were enclosed in aluminum pans and purged with nitrogen at 10 mL/min. Initially, they were heated from ambient temperature to 220 ℃, erasing thermal history. They were then cooled to -90 ℃ and reheated to 220 ℃, with all tests conducted at 10 ℃/min.
2.3.5. X-ray diffraction (XRD). XRD test (Bruker, D8) was carried out to characterize the crystallization of elastomer.
2.3.6. Polarized Optical Microscope (POM). For POM studies, BPU samples (50 mg/mL, DMF) were cast into PTFE molds. Controlled evaporation yielded films (100 μm) displaying spherulitic structures. The equipment utilized was a Nanoscope IV, Dimension 3100 from Digital Instruments.
2.3.7. Tensile Experiments. Specimens (1×2×12 mm, n = 5) of each PU were prepared and stretched to failure at a rate of 10 mm/min using an Instron 5966 uniaxial tensile tester equipped with a 1 kN load cell. The elastic modulus, tensile strength, and ultimate elongation at break were calculated from the resultant engineering stress/strain curves. A secant modulus based on 2% strain was calculated for the elastic modulus and subsequently referred to as the modulus.
2.3.8. In Vitro Degradation Study. In vitro degradation tests for the samples were conducted in 0.025 M phosphate-buffered saline (PBS; pH 7.4) at 37 ℃. Disk-shaped polymeric specimens, approximately 4.5 mm in diameter and 1 mm thick, were extracted from a compression-molded sheet. These specimens were submerged in tubes containing PBS, both enzyme-free (10 mL) and with P. lipase (10 mL, 1 mg/mL). They were then placed in an incubator shaker at 100 rpm and 37 °C. The PBS was refreshed every 3 days. After a set duration, samples were retrieved, rinsed, and vacuum-dried at ambient temperature until a consistent weight was achieved for evaluation. The weight loss percentage was determined as:
% weight loss = \( \frac{Mi-Mf}{Mi} \) × 100
where Mi represents the sample’s initial weight and Mf denotes its constant weight post-degradation at varying time intervals with PBS hydrolysis.
2.3.9. In Vitro Cytotoxicity. To measure the viability of myoblast cells on the synthesized polymeric samples, an MTT assay was conducted. Cells were cultured on sterilized samples in 24-well plates at a density of 3 × 10^3 cells/well and incubated for 5 days in a 5% CO2 environment at 37 °C with 95% humidity. After incubation, the medium was replaced with 0.5 mg of MTT/mL reagent in DMEM without serum. The enzyme mitochondrial succinate dehydrogenase in active cells converts the yellow MTT to blue-violet formazan crystals. This conversion rate corresponds to the number of viable cells on the sample. Post 3-hour incubation at 37 °C, excess MTT was discarded, and formazan crystals were dissolved using 200 μL of DMSO. The optical density (OD) was measured using an ELISA reader (iMark, Biorad Laboratories, India) at 595 nm. Viability percentage was also determined as follows:
% cell viability = \( \frac{OD of the sample}{OD of the control} \) × 100
3. Results and Discussion
3.1. Synthesis Results of BPU
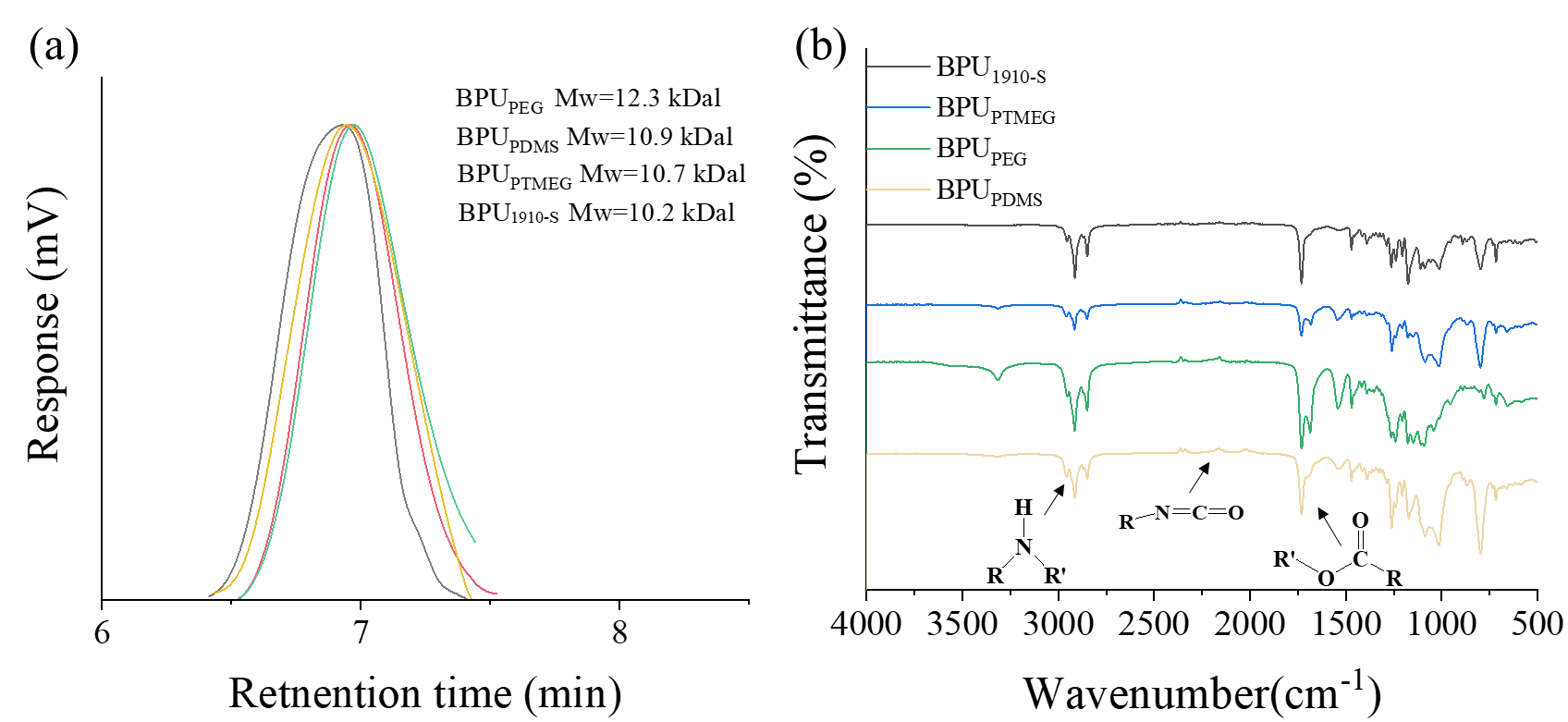
Figure 2. Characterization of BPUs using GPC and ATR-IR techniques. (a) GPC charts of BPUPTMEG, BPUPDMS, BPUPEG, and BPU1910-S with DMF as the eluent phase. (b) ATR-IR spectra in the range from 1500 to 3500 cm-1 for the BPUs.
Figure 2(a) is the results of the GPC test for four different samples: BPUPTMEG, BPUPDMS, BPUPEG and BPU1910-s. As shown in Figure 2(a), BPUPTMEG has a molecular weight for about 12.3 kDal; BPUPDMS has a molecular weight for about 10.9 kDal; BPUPEG has molecular weight for about 10.7 kDal; BPU1910-S has a molecular weight for about10.2 kDal. All of the datas are around 10 kDal, which indicates the synthesis process gains success. Besides, the PDI for all four samples are around 2.32. The similarities on PDI and molecular weight make sure that these two do not generate influence on the difference between mechanical or other properties of the BPU. AFR-IR spectras of the BPU samples are depicted in Figure 2(b), spanning wavenumbers from 1500 to 3500 cm-1. Notably, the absence of a stretching vibration near 2270 cm-1 confirms the lack of residual isocyanate groups across all BPU samples, suggesting a complete reaction during synthesis. The presence of stretching vibrations within the 3200-3500 cm-1 and 1655-1760 cm-1 ranges further confirms the existence of -NH and -C=O functional groups, respectively, underscoring the successful synthesis of the BPU samples.
3.2. Analysis of BPU Structure
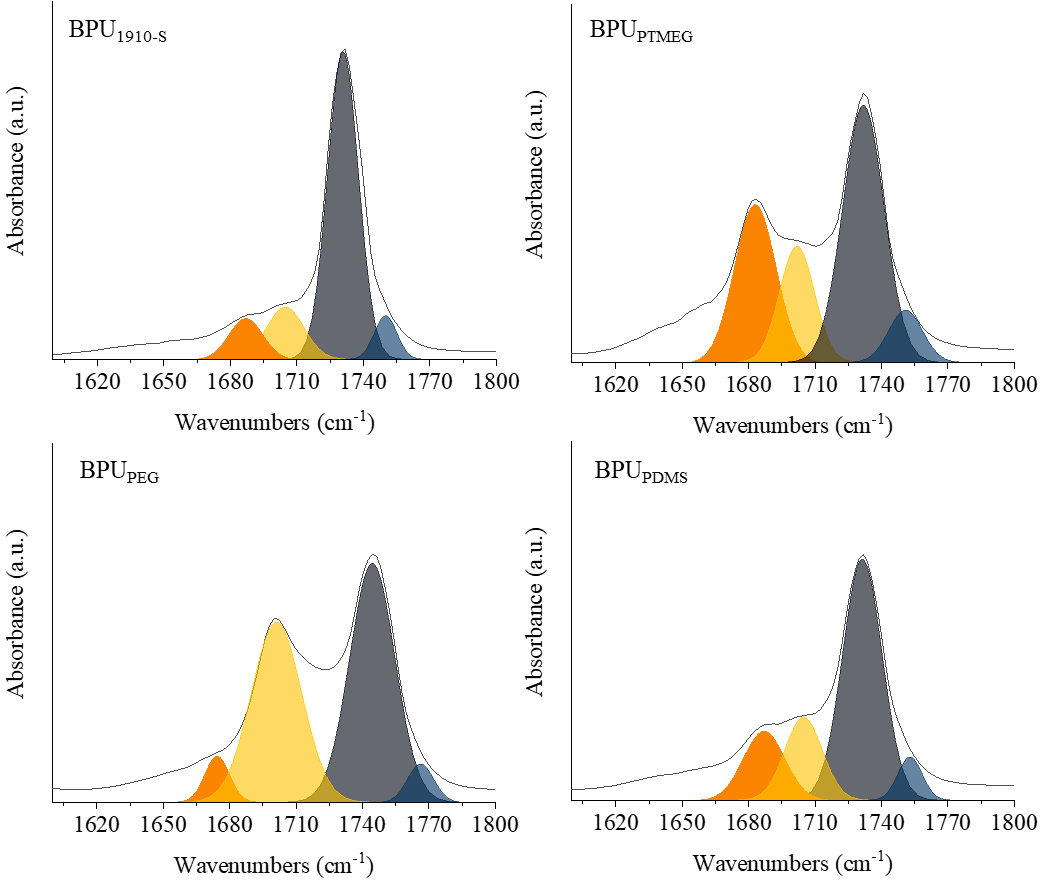
Figure 3. Analysis of -C=O group absorption in various morphological structures. ATR-IR spectra in the range of 1655 - 1775 cm-1 with signals attributed to the absorption peak of -C=O groups.
The ATR-IR spectra, particularly between 1655 to 1775 cm-1, were examined to identify signals from four unique -C=O groups. These include: 1. Free carbonyl groups at 1732-1775 cm-1 (Band I).2. Hydrogen-bonded -C=O at the hard/soft domain interface at 1695-1760 cm-1 (Band II).3. Hydrogen-bonded -C=O in ordered hard domains at 1680-1735 cm-1 (Band III).4. Hydrogen-bonded -C=O in the polyester polyol of soft domains at 1655-1710 cm–1 (Band IV). (shown in Figure 3)16. Upon comparison with BPU1910-S, which possesses pure castor oil soft chain segments, BPUPEG’s absorption peak at Band II reveals a heightened area percentage. Conversely, its absorption peak at Band III displays a diminished area percentage, suggesting a more structured presence of hard segments in BPUPEG. The absorption peak of BPUPTMEG at Band II exhibits a marked increase in area percentage, while its Band III peak shows a significant decrease. This trend is attributed to the declining crystallinity of the B block, which weakens its molecular chain binding, thereby reducing BPUPTMEG’s hard segment regularity and resulting in a more relaxed system structure (shown in SI Table S1).
Interestingly, despite PDMS having the lowest crystallinity, BPUPDMS’s absorption peak at Band III, when compared to BPUPTMEG, shows an increase. This anomaly arises from the pronounced thermodynamic incompatibility between PU hard segments and PDMS. Spatial extrusion fosters an increase in the ordered structure of hard segments. However, the percentage of the ordered structure induced by spatial extrusion remains limited, proving insufficient to substantially enhance the mechanical attributes of BPU.
These findings underscore the pivotal role of introducing A-B-A triblock polyols into BPU as select soft segments. The B block, with its superior crystallinity, can restrict the mobility of BPU molecular chains. This limitation mitigates the structural disruption caused by biological polyols, thereby bolstering the orderliness of PU hard segments.
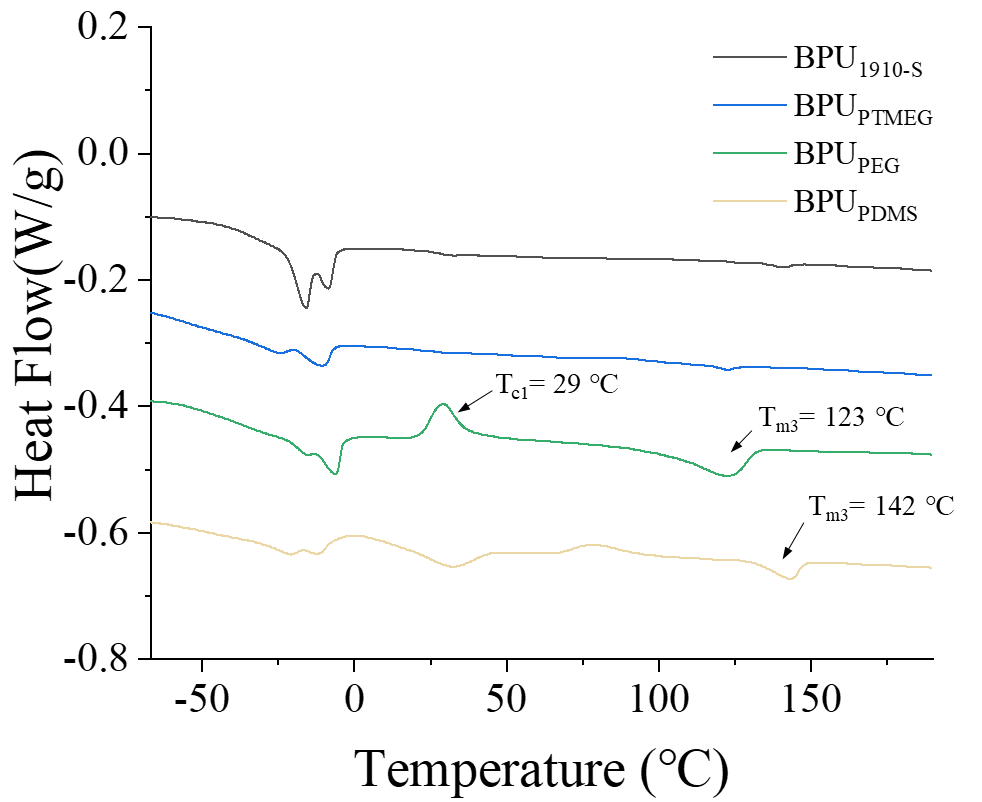
Figure 4. Thermal analysis of BPUs using DSC. DSC spectra of BPUPTMEG, BPUPDMS, BPUPEG, and BPU1910-S with temperature ranging from -90 to 220 ℃.
Figure 4 delineates the DSC results for the four BPU samples, with temperatures spanning from -90 to 220 ℃. Notably, among the quartet, only BPUPEG exhibits a crystallized peak at 29 ℃, attributable to the inherent crystallinity of the PEG block8. The PEG block, renowned for its pronounced crystallinity, can undergo crystallization at ambient temperatures, a feat unattainable by other B blocks within the A-B-A triblock framework.
Furthermore, both BPUPDMS and BPUPEG manifest a melting peak at elevated temperatures, indicative of the hard segments within these samples. In contrast, BPUPTMEG and BPU1910-S, owing to the disarray within their hard segments and the perturbations introduced by branching in the BPU, lack a discernible melting peak.
The mechanisms underpinning the orderly arrangement of hard segments in BPUPEG and BPUPDMS diverge. In the case of BPUPDMS, the pronounced thermodynamic incompatibility between PDMS and its hard segment results in spatial repulsion between the two. Consequently, hard segments are spatially constrained, fostering a more organized arrangement. For BPUPEG, the PEG block’s robust crystallinity bolsters the structural stability of its hard segment, mitigating the disruptions posed by the branched chains of bio-based polyols. Additionally, the PCL, employed as a linker between the block and BPUPEG, further augments structural stability, rendering the hard segment of BPUPEG more regimented. Collectively, these findings underscore the efficacy of our approach in modulating the ordered hard segment within BPUPEG.

Figure 5. Microscopic and structural analysis of BPUs. (a) POM images of BPUPTMEG, BPUPDMS, BPUPEG, and BPU1910-S. (b) WAXS spectra of BPUPTMEG, BPUPDMS, BPUPEG, and BPU1910-S.
Figure 5(a) presents the POM results, with arrows highlighting the hard segment crystal structures of BPU. Notably, the hard segment crystal structure of BPU1910-S appears somewhat dispersed. This is attributed to the branched chain of 1910-S disrupting the hydrogen bond interactions within the hard segment, thereby diminishing its organized arrangement. The crystalline structure dimensions for BPUPEG, BPUPTMEG, and BPUPDMS are 4 microns, 7 microns, and 12 microns, respectively. The compactness of BPUPEG’s structure arises from the PEG block crystallization, which curtails molecular chain mobility and minimizes the structural perturbations caused by the branched chains of bio-based polyols. Furthermore, the PCL block refines the structure of the BPUPEG hard segment, facilitating its even distribution within the BPUPEG system. These POM findings validate our strategic approach: employing PCL-PEG-PCL polyol as the soft segment in BPUPEG ensures that the PCL can standardize the hard segment’s dimensions, achieving a consistent distribution. Concurrently, the crystalline nature of the PEG block augments the sample’s rigidity.
To discern the nanocrystalline structures within the BPU samples, WAXS analyses were conducted and the results are shown in Figure 5(b). BPUPEG exhibited a distinct crystalline peak at 21.5°, characteristic of both the BPU hard segment and PEG. Compared to its counterparts, the crystalline peak of BPUPEG at 21.5° is markedly more pronounced and intense. As the crystallinity of the B block in the A-B-A triblock polyol diminishes, the peaks for BPUPTMEG and BPUPDMS at 21.5° become increasingly subdued. This trend underscores the presence of crystalline PEG blocks within the BPUPEG system and the enhanced orderliness of its hard segment. A decline in the crystallinity of the B block in the A-B-A triblock copolymer results in a more disorganized hard segment arrangement in BPUPEG, BPUPTMEG, and BPU1910-S. This, in turn, reduces crystallization levels, leading to the attenuation of the BPU’s crystalline peak at 21.5°.
3.3. Performance of BPU Properties
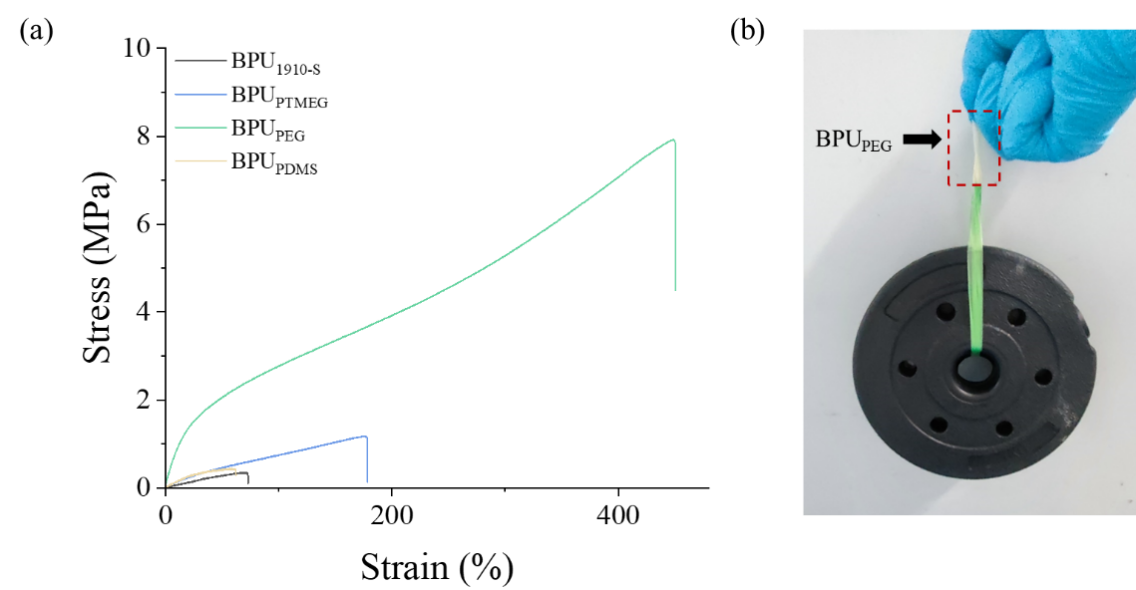
Figure 6. Mechanical properties and demonstration of BPUs. (a) Stress-strain curves of BPUPTMEG, BPUPDMS, BPUPEG, and BPU1910-S. (b) Photographs showcasing the strength of BPUPEG by holding two 1kg dumbbell pieces.
The results of the stress-strain test are shown in Figure 6. It can be observed from Figure 6(a) that the breaking stress of BPUPEG is 7.9Mpa, and the fracture strain is 447%. In comparison to BPU1910-S, BPUPEG’s mechanical attributes have seen a substantial enhancement. Specifically, the fracture stress has surged from 0.4 MPa to 7.9 MPa, and the fracture strain has escalated from 62% to 447%. This improvement correlates with the heightened crystallinity of the B block in the A-B-A triblock polyol observed in other BPU samples (shown in SI Table S2). Figure 6(b) shows that the mechanical performance of BPUPEG is good enough to hold two 1kg dumbbell pieces.
Among the samples tested, BPUPEG stands out with its superior mechanical properties. This can be attributed to the reinforcing effect of the PEG crystalline phase in BPUPEG, which also functions as a physical cross-linking juncture. Furthermore, the uniform distribution and optimal sizing of hard segments in BPUPEG contribute to its robustness. The combined resistance offered by the PEG crystalline phase and the organized hard segments in BPUPEG effectively counter external stresses, thereby amplifying its mechanical resilience.
However, as the crystallinity of the B block in the A-B-A triblock polyol diminishes, there’s a discernible decline in the orderly arrangement of hard segments across BPUPEG, BPUPTMEG, and BPU1910-S. The diminishing role of the B block as a physical cross-linking nexus results in the tapering mechanical properties of BPUPTMEG and BPUPDMS.
In essence, the robust mechanical attributes of BPUPEG underscore the efficacy of our design approach. By integrating A-B-A triblock polyols into BPU, where block A is represented by PCL (modulating the hard segment size of BPU), and adopting a highly crystalline PEG block, we’ve achieved a dual benefit. The PEG block not only serves as an interlinking bridge between blocks but also fortifies molecular chains. This minimizes the perturbations caused by the 1910-S branching to the BPU hard segments, enhancing their organization. Consequently, this intricate mechanism bolsters the mechanical prowess of BPUPEG.
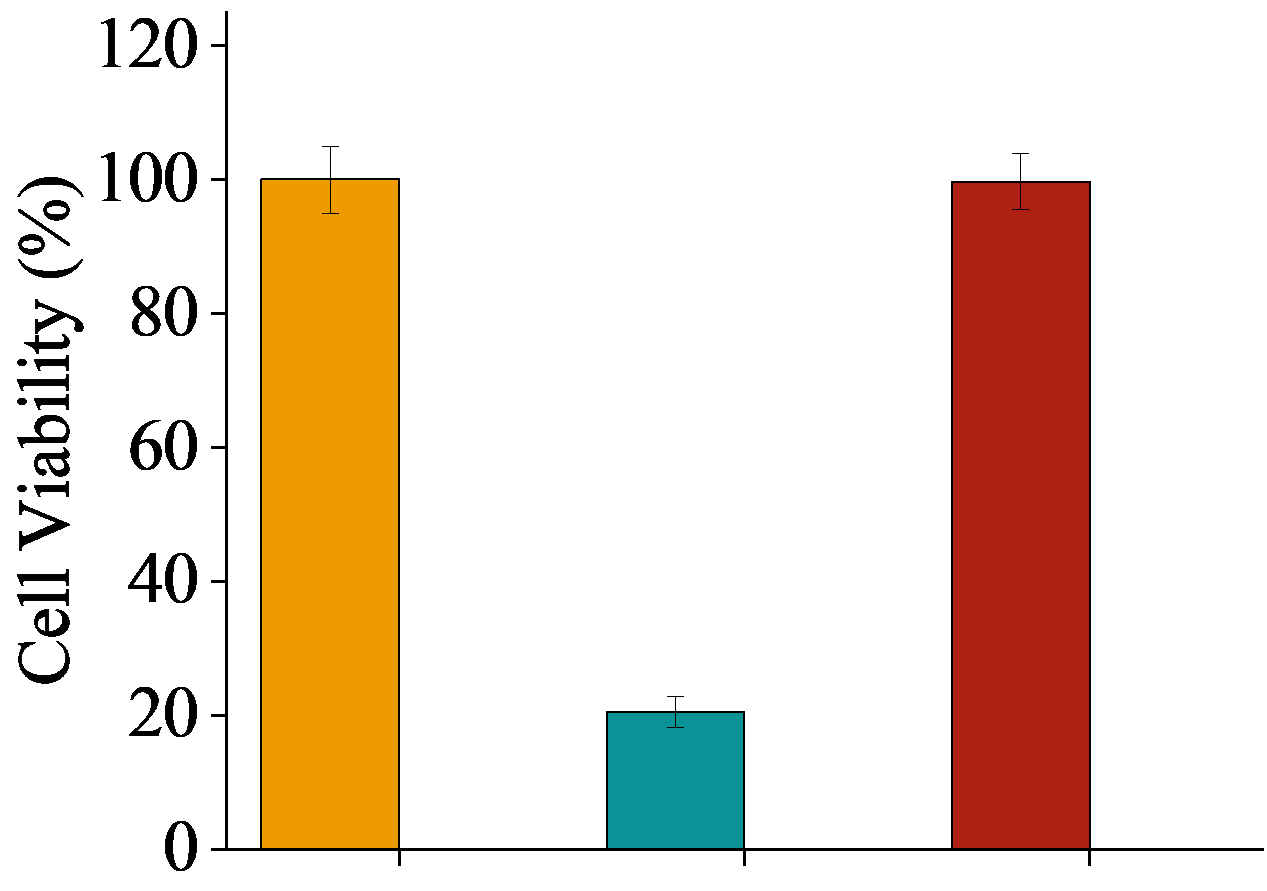
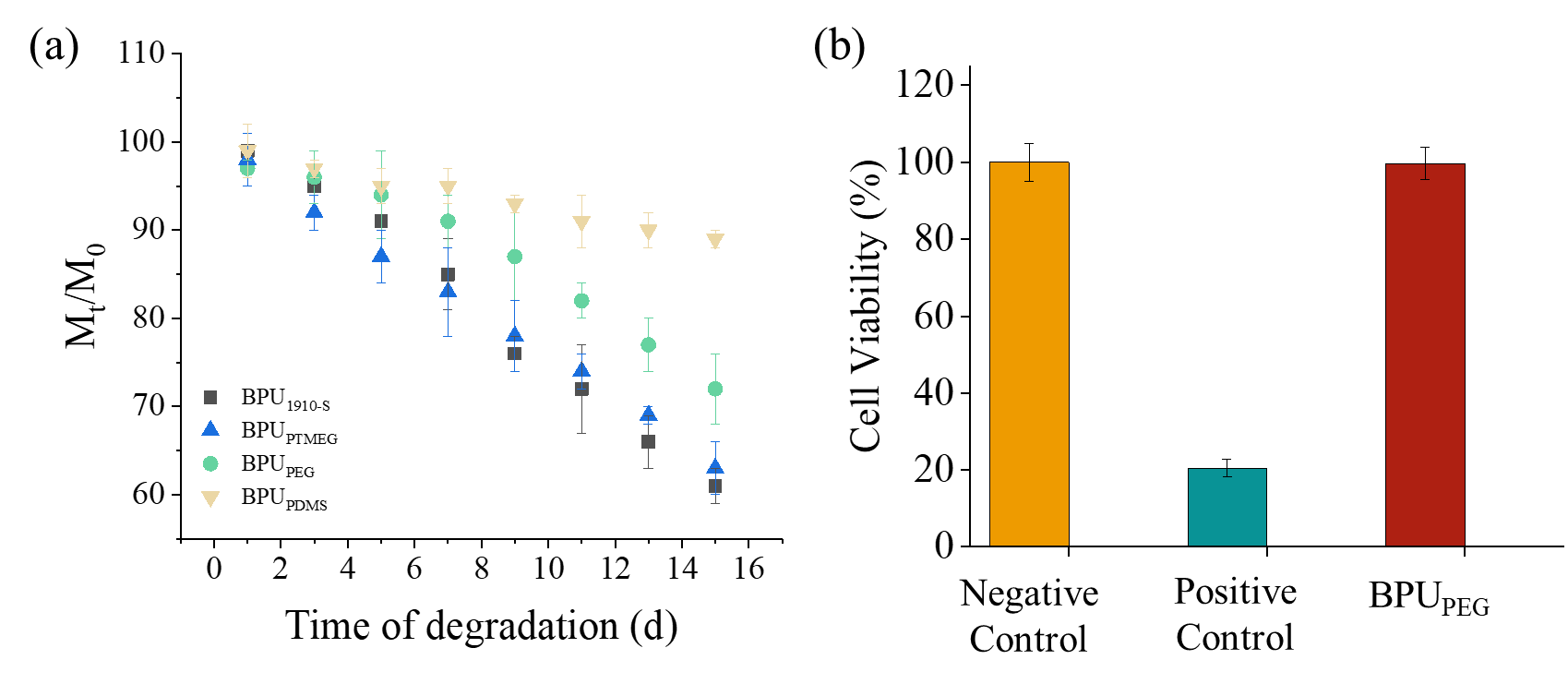
Figure 7. Degradation analysis and biocompatibility assessment of BPUs. (a) Variation of the weight loss as a function of the degradation time for BPUPTMEG, BPUPDMS, BPUPEG, and BPU1910-S. (b) MTT assay plot illustrating the proliferation of HE cells in the presence of various BPU hydrolyzed extracts.
Figure 7 presents the biodegradation results for the BPU samples. As anticipated, all BPUs exhibited biodegradable characteristics. However, the biodegradation rates among the four BPUs were distinct. BPUPTMEG and BPU1910-S showcased the most rapid biodegradation rates, retaining merely 60% of their original weight after a 14-day hydrolysis period. In contrast, BPUPEG demonstrated the most gradual hydrolysis rate, preserving 90% of its initial weight over the same duration.
The accelerated hydrolysis observed in BPUPTMEG and BPU1910-S can be attributed to their less compact structures, facilitating the easy penetration of water molecules and thereby hastening the hydrolysis process. Conversely, the crystalline phase of PEG in BPUPEG acts as a barrier, limiting the ingress of water molecules and subsequently decelerating the hydrolysis rate. The PDMS block in BPUPDMS, being highly hydrophobic, offers resistance against the hydrolytic action on the PCL blocks, significantly diminishing the hydrolysis rate.
To assess the potential toxicity of the degradation products, the hydrolysate from BPUPEG underwent MTT testing. The results were encouraging, revealing negligible cytotoxicity with a cell survival rate of 99.8%.
In conclusion, all four BPUs manifest commendable biodegradability. Notably, the degradation byproducts of BPUPEG are non-toxic and can be safely released into the environment. This underscores BPUPEG’s potential as an environmentally friendly material, aligning with future sustainability goals centered on carbon conservation and emission reduction.

Figure 8. Recycling methodology and mechanical comparison of the elastomer. (a) Recycling process for the elastomer: i) Segmentation of the elastomer. ii) Dissolution in DMF and spreading on a Petri dish. iii) Subsequent recycling of the elastomer. (b) Stress-strain graphs comparing the mechanical properties of the original and recycled elastomer after the initial cutting/recycling cycle.
The recyclability of BPU was evaluated by dissecting the elastomer and subsequently dissolving it in DMF. The stress-strain data post the initial cycle is depicted in Figure 8(b). As observed from the experimental results in Figure 8(a), BPUPEG fragments, when dissolved in DMF, can be reconstituted into a diaphragm by recasting them into the mold. Further tensile tests, as presented in Figure 8(b), revealed that the mechanical attributes of the reformed BPUPEG closely mirrored those of the pristine material. This inherent recyclability stems from BPUPEG’s classification as a thermoplastic BPU, which allows it to be solubilized in organic solvents like DMF. Upon the evaporation of the solvent, BPUPEG can be reconstituted, retaining its mechanical integrity throughout the recycling process.
In essence, BPUPEG not only boasts superior mechanical and biodegradable properties but also exemplifies recyclability. This positions it as a prime candidate for BPU materials, aligning with future sustainability objectives centered on carbon conservation and emission mitigation.
4. Conclusion
In the realm of sustainable materials, BPUs have emerged as a promising alternative to their petroleum-based counterparts. This study, centered on BPUPEG, has illuminated its potential as a frontrunner in this category.
Our investigations, through GPC and ATR-IR analyses, confirmed the unique molecular structure and uniformity of BPUPEG. This uniformity is pivotal, translating directly to its enhanced mechanical properties. DSC, POM, and WAXS tests further highlighted BPUPEG’s crystalline nature, with the PEG block’s crystallinity playing a dual role: imparting strength and acting as a physical cross-linking point.
Mechanical resilience, however, is just one facet of BPUPEG’s appeal. Our biodegradation experiments showcased its commendable environmental compatibility, ensuring that post-functional life, BPUPEG can reintegrate into the environment without adverse effects. Adding to its eco-friendly credentials is its recyclability. BPUPEG can be dissolved, reformed, and reused without compromising its inherent properties, emphasizing its potential for sustainable applications.
Furthermore, the hydrolysate tests vouched for the non-toxic nature of BPUPEG’s degradation products, an essential attribute for any material intended for broader applications.
Looking ahead, the application prospects for BPUPEG are vast and varied. Given its unique combination of mechanical strength, biodegradability, and recyclability, BPUPEG is poised to revolutionize industries ranging from automotive to medical. Its potential in the packaging sector, where the demand for sustainable yet durable materials is ever-growing, is particularly noteworthy. In the medical field, BPUPEG’s biocompatibility could lead to its use in implants or drug delivery systems. Moreover, in sectors like construction and textiles, BPUPEG’s resilience and environmental friendliness could redefine material standards
In essence, BPUPEG stands out not just for its mechanical prowess but also for its environmental compatibility. It embodies the future of sustainable materials, balancing performance with eco-friendliness, and sets a benchmark for future research in the domain of bio-based materials.
5. Supplementary Materials: Characterization Methods
5.1. Nuclear Magnetic Resonance Spectroscopy (NMR)
High resolution 1H-NMR spectra of HO-PCL-PEG-PCL-OH, HO-PCL-PTMEG-PCL-OH and HO-PCL-PDMS-PCL-OH were recorded using a Bruker 400 MHz spectrometer, respectively. The samples were dissolved in deuterated chloroform (CDCl3) at a concentration of up to 10 wt.%. Chemical shifts were referenced to tetramethylsilane (TMS) as the standard.
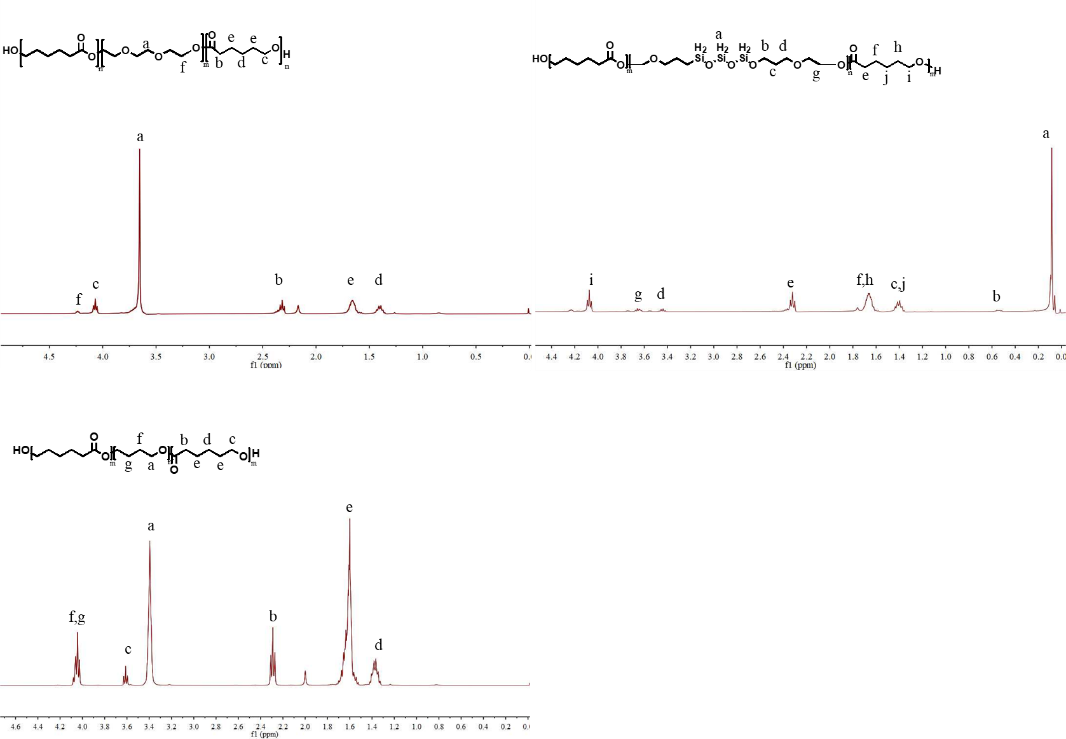
Figure S1. 1H-NMR spectra of the tri-block co-polymer diols of HO-PCL-PEG-PCL-OH, HO-PCL-PTMEG-PCL-OH and HO-PCL-PDMS-PCL-OH in CDCl3 at room temperature.
5.2. Gel Permeation Chromatography (GPC)
GPC (Agilent/Wyatt 1260) was used to determine the molecular weights (Mw.) and PDI of the polyurethanes at 30 °C. Anhydrous N,N-Dimethylformamide (DMF) for 1910-S, HO-PCL-PTMEG-PCL-OH and HO-PCL-PEG-PCL-OH, anhydrous Tetrahydrofuran (THF) for HO-PCL-PDMS-PCL-OH were used as the elution phase at a flow rate of 1.0 mL/min, respectively. Narrow polyethylene oxide standards with molecular weights ranging from 550 to 100,000 g/mol were used to calibrate the system.
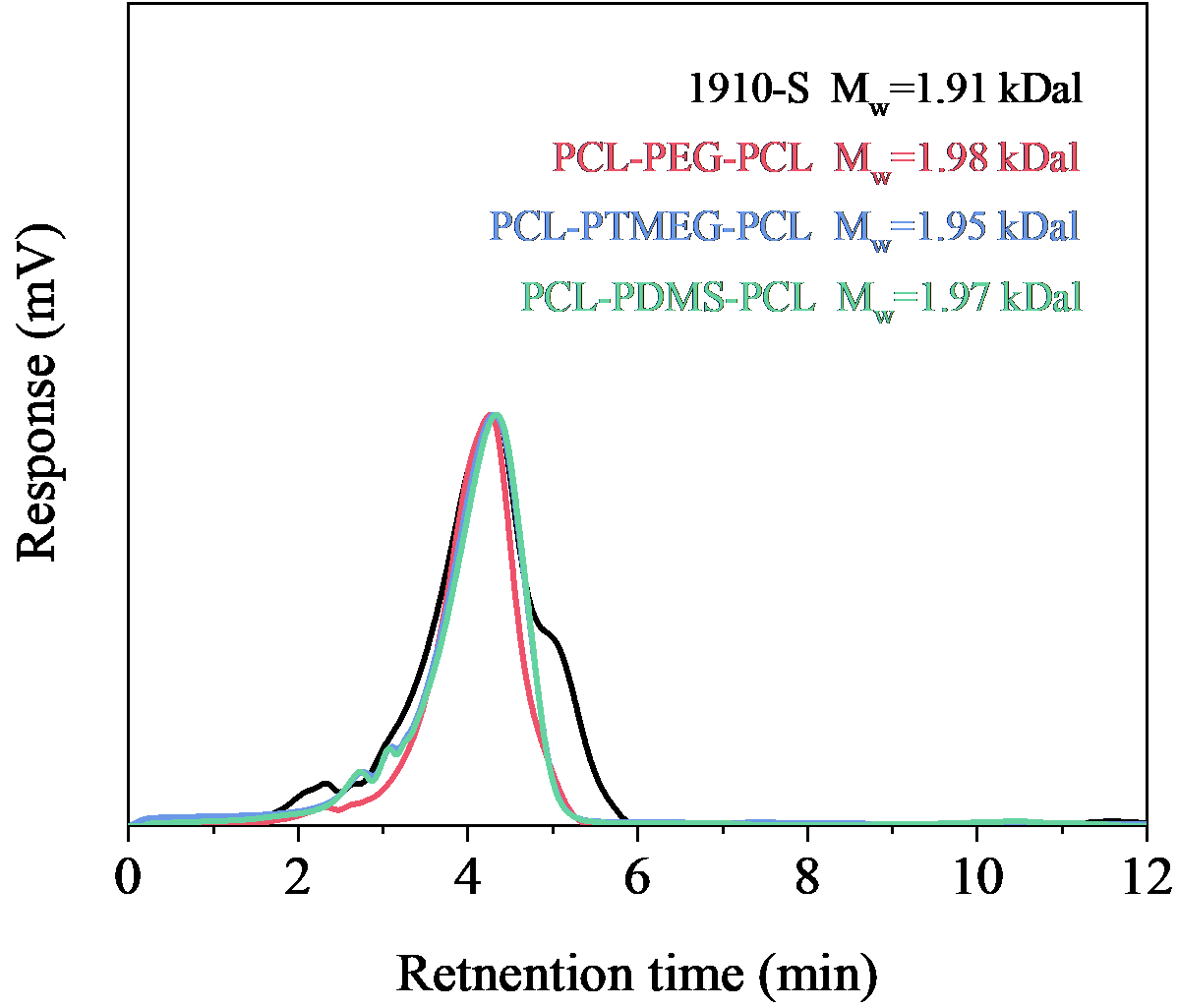
Figure S2. GPC charts of the macromolecular initiators and the corresponding tri-block co-polymer diols in DMF or THF.
5.3. Attenuated Total internal Reflection Infrared Spectroscopy (ATR-IR)
In the ATR-IR spectra, non-hydrogen-bonded (free) carbonyl groups appear at 1732 - 1770 cm-1 (Band I), hydrogen-bonded carbonyl groups in disordered conformations appears at 1695 - 1760 cm-1 (Band II), hydrogen-bonded carbonyl groups in ordered hard domains can be observed at 1680 - 1735 cm-1 (Band III) and hydrogen-bonded carbonyl groups in polyester polyol in ordered soft domains appears at 1655-1710 cm-1 (Band IV).1 The different bands in the ATR-IR spectra were assigned and the area percentages (area%) of the corresponding bands were calculated and summarized in Table S1.
Table S1. The calculated area percentages (area%) of the corresponding bands in the ATR-IR spectra for each of the BPU.
Sample | Band I (% area ~1750cm-1 ) | Band II (% area ~1730cm-1 ) | Band III (% area ~1700cm-1 ) | Band IV (% area ~1690cm-1 ) |
BPUPTMEG | 7.7 | 46.2 | 17.5 | 28.6 |
BPUPDMS | 4.8 | 58.4 | 19.2 | 17.6 |
BPUPEG | 4.5 | 49.1 | 41.3 | 5.1 |
BPU1910-S | 7.1 | 68.5 | 14.2 | 10.2 |
5.4. Tensile Experiments
Table S2. Summary of the tensile stress and elongation at break for each of the BPU (n = 5).
Sample | Stress at break (Mpa) | Stress at break (%) |
BPUPTMEG | 1.2±0.2 | 176±17 |
BPUPDMS | 0.4±0.1 | 62±8 |
BPUPEG | 7.9±0.4 | 447±21 |
BPU1910-S | 0.33±0.1 | 72±11 |
Acknowledgments
Authors wishing to acknowledge assistance or encouragement from colleagues, special work by technical staff or financial support from organizations should do so in an unnumbered Acknowledgments section immediately following the last numbered section of the paper.
References
[1]. Engels H W, Pirkl H G, Albers R, et al. Polyurethanes: Versatile materials and sustainable problem solvers for today’s challenges[J]. Angewandte Chemie International Edition, 2013,52(36):9422-9441.
[2]. Zhao D, Liang X, Wang J, et al. Vanillin-Based Degradable Polyurethane Thermosets Demonstrating High Bio-Content and Mechanical Properties[J]. ACS Applied Polymer Materials, 2023.
[3]. Zhang C, Kessler M R. Bio-based polyurethane foam made from compatible blends of vegetable-oil-based polyol and petroleum-based polyol[J]. ACS Sustainable Chemistry & Engineering, 2015,3(4):743-749.
[4]. Zhang C, Madbouly S A, Kessler M R. Biobased polyurethanes prepared from different vegetable oils[J]. ACS applied materials & interfaces, 2015,7(2):1226-1233.
[5]. Tenorio-Alfonso A, Sánchez M C, Franco J M. A review of the sustainable approaches in the production of bio-based polyurethanes and their applications in the adhesive field[J]. Journal of Polymers and the Environment, 2020,28:749-774.
[6]. Schneiderman D K, Vanderlaan M E, Mannion A M, et al. Chemically recyclable biobased polyurethanes[J]. ACS Macro Letters, 2016,5(4):515-518.
[7]. Liu L, Lu J, Zhang Y, et al. Thermosetting polyurethanes prepared with the aid of a fully bio-based emulsifier with high bio-content, high solid content, and superior mechanical properties[J]. Green Chemistry, 2019,21(3):526-537.
[8]. Kong W, Lei Y, Jiang Y, et al. Preparation and thermal performance of polyurethane/PEG as novel form-stable phase change materials for thermal energy storage[J]. Journal of Thermal Analysis and Calorimetry, 2017,130:1011-1019.
[9]. Wang X, Zhan S, Lu Z, et al. Healable, Recyclable, and Mechanically Tough Polyurethane Elastomers with Exceptional Damage Tolerance[J]. Advanced Materials, 2020,32(50):2005759.
[10]. Calvo-Correas T, Martin M D, Retegi A, et al. Synthesis and Characterization of Polyurethanes with High Renewable Carbon Content and Tailored Properties[J]. ACS Sustainable Chemistry & Engineering, 2016,4(10):5684-5692.
[11]. Lee S, Yuk J S, Park H, et al. Multiblock Thermoplastic Elastomers Derived from Biodiesel, Poly(propylene glycol), and l-Lactide[J]. ACS Sustainable Chemistry & Engineering, 2017,5(9):8148-8160.
[12]. Laurichesse S, Avérous L. Chemical modification of lignins: Towards biobased polymers[J]. Progress in Polymer Science, 2014,39(7):1266-1290.
[13]. Shi J, Zheng T, Zhang Y, et al. Reprocessable Cross-Linked Polyurethane with Dynamic and Tunable Phenol–Carbamate Network[J]. ACS Sustainable Chemistry & Engineering, 2020,8(2):1207-1218.
[14]. Zhang C, Kessler M R. Bio-based Polyurethane Foam Made from Compatible Blends of Vegetable-Oil-based Polyol and Petroleum-based Polyol[J]. ACS Sustainable Chemistry & Engineering, 2015,3:743-749.
[15]. Gang H, Lee D, Choi K, et al. Development of High Performance Polyurethane Elastomers Using Vanillin-Based Green Polyol Chain Extender Originating from Lignocellulosic Biomass[J]. ACS Sustainable Chemistry & Engineering, 2017,5(6):4582-4588.
[16]. Ma J, Deng B, Fan Y, et al. Polyurethane elastomers with amphiphilic ABA tri-block co-polymers as the soft segments showing record-high tensile strength and simultaneously increased ductility[J]. Polymer Chemistry, 2022,13(35):5159-5168.
Cite this article
Wu,J.;Zheng,B. (2024). Recyclable and biodegradable bio-based polyurethane elastomers with exceptional mechanically properties. Applied and Computational Engineering,58,194-207.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Materials Chemistry and Environmental Engineering
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Engels H W, Pirkl H G, Albers R, et al. Polyurethanes: Versatile materials and sustainable problem solvers for today’s challenges[J]. Angewandte Chemie International Edition, 2013,52(36):9422-9441.
[2]. Zhao D, Liang X, Wang J, et al. Vanillin-Based Degradable Polyurethane Thermosets Demonstrating High Bio-Content and Mechanical Properties[J]. ACS Applied Polymer Materials, 2023.
[3]. Zhang C, Kessler M R. Bio-based polyurethane foam made from compatible blends of vegetable-oil-based polyol and petroleum-based polyol[J]. ACS Sustainable Chemistry & Engineering, 2015,3(4):743-749.
[4]. Zhang C, Madbouly S A, Kessler M R. Biobased polyurethanes prepared from different vegetable oils[J]. ACS applied materials & interfaces, 2015,7(2):1226-1233.
[5]. Tenorio-Alfonso A, Sánchez M C, Franco J M. A review of the sustainable approaches in the production of bio-based polyurethanes and their applications in the adhesive field[J]. Journal of Polymers and the Environment, 2020,28:749-774.
[6]. Schneiderman D K, Vanderlaan M E, Mannion A M, et al. Chemically recyclable biobased polyurethanes[J]. ACS Macro Letters, 2016,5(4):515-518.
[7]. Liu L, Lu J, Zhang Y, et al. Thermosetting polyurethanes prepared with the aid of a fully bio-based emulsifier with high bio-content, high solid content, and superior mechanical properties[J]. Green Chemistry, 2019,21(3):526-537.
[8]. Kong W, Lei Y, Jiang Y, et al. Preparation and thermal performance of polyurethane/PEG as novel form-stable phase change materials for thermal energy storage[J]. Journal of Thermal Analysis and Calorimetry, 2017,130:1011-1019.
[9]. Wang X, Zhan S, Lu Z, et al. Healable, Recyclable, and Mechanically Tough Polyurethane Elastomers with Exceptional Damage Tolerance[J]. Advanced Materials, 2020,32(50):2005759.
[10]. Calvo-Correas T, Martin M D, Retegi A, et al. Synthesis and Characterization of Polyurethanes with High Renewable Carbon Content and Tailored Properties[J]. ACS Sustainable Chemistry & Engineering, 2016,4(10):5684-5692.
[11]. Lee S, Yuk J S, Park H, et al. Multiblock Thermoplastic Elastomers Derived from Biodiesel, Poly(propylene glycol), and l-Lactide[J]. ACS Sustainable Chemistry & Engineering, 2017,5(9):8148-8160.
[12]. Laurichesse S, Avérous L. Chemical modification of lignins: Towards biobased polymers[J]. Progress in Polymer Science, 2014,39(7):1266-1290.
[13]. Shi J, Zheng T, Zhang Y, et al. Reprocessable Cross-Linked Polyurethane with Dynamic and Tunable Phenol–Carbamate Network[J]. ACS Sustainable Chemistry & Engineering, 2020,8(2):1207-1218.
[14]. Zhang C, Kessler M R. Bio-based Polyurethane Foam Made from Compatible Blends of Vegetable-Oil-based Polyol and Petroleum-based Polyol[J]. ACS Sustainable Chemistry & Engineering, 2015,3:743-749.
[15]. Gang H, Lee D, Choi K, et al. Development of High Performance Polyurethane Elastomers Using Vanillin-Based Green Polyol Chain Extender Originating from Lignocellulosic Biomass[J]. ACS Sustainable Chemistry & Engineering, 2017,5(6):4582-4588.
[16]. Ma J, Deng B, Fan Y, et al. Polyurethane elastomers with amphiphilic ABA tri-block co-polymers as the soft segments showing record-high tensile strength and simultaneously increased ductility[J]. Polymer Chemistry, 2022,13(35):5159-5168.