







1. Introduction
Continuous development of human society demands significant energy, and currently mainly from fossil fuels, leading to the significant greenhouse gas emissions, global warming and the energy-environment crisis [1]. Hence, to achieve the sustainable development of our society, a paradigm shift in the energy landscape is needed, necessitating the development and use of sustainable and renewable resources with less, zero and/or negative carbon emissions. Hydrogen has been recognized as one of the ultimate green energies, yet its current production is associated with considerable carbon footprints (e.g., via steam reforming of hydrocarbons, mainly methane, from fossil fuels) [2]. Conversely, hydrogen from water electrolysis, when green electricity from renewable sources (such as solar and wind) is employed, could be the solution for producing green hydrogen with significantly reduced carbon emissions [3]. The technology mimics photosynthesis in nature powered by electricity to split water into hydrogen and oxygen, in which catalysts are needed to improve the process efficiency. However, the process efficiency of the current development is not satisfactory which demands further design and optimization of relevant catalysts to achieve better efficiency and economy.
The common state-of-the-art catalysts for water splitting processes are that based on noble metals, such as Ir and Ru, which are comparatively expensive and scarce. Comparatively, non-noble metals such as Fe, Co, Ni, and Cu could be more ideal, though their metal centers need to be engineered to improve the process efficiency of artificial water oxidation. It is recognized that the oxygen evolution process in photosynthesis is highly analogous to artificial water oxidation [4]. The oxygen evolution rate in natural photosynthesis ranges from 100 to 400 s-1, hence, it is challenging to achieve such a good efficiency with non-noble metal catalysts. The high efficiency of oxygen evolution in photosynthesis is attributed to the participation of multiple catalytic cores. Therefore, introducing multi-core synergism for microscopic adjustments is an effective means to achieve this enhancement. Given that the metal center is key to catalytic activity, the development of catalysts with multi-active sites becomes particularly important.
This mini-review focuses on summarizing the most recent studies in the fields of dual-site catalysis, via comparative metanalysis of the state-of-the-arts, we provide critical comments on the current developments, as well as providing our perspective on the fields. The review is a timely snapshot of the field, establishing the current boundary of the technical development for progressing the research to the next stage.
2. Advances in Water Oxidation Catalysis
2.1. Ruthenium-based Catalysts
Ruthenium (Ru) is a noble metal commonly explored in water oxidation catalysis, holding a significant position in energy conversion and storage technologies due to its high efficiency, stability, and tunability. Ru-based catalysts typically exhibit high catalytic activity in water oxidation reactions, for example, Ru-bda based catalysts in Figure 1 (bda = 2,2′-bipyridine-6,6′-dicarboxylate) are the earliest catalysts that can match the rate of water oxidation in natural photosynthesis, achieved a turnover frequency (TOFs) over 300 s−1, facilitating O–O bond formation via the interaction of two M−O units (I2M) pathway (Figure 1). Supramolecular interactions between the catalysts serve as the primary driving force for their dimerization, with the pyridine axial ligand in Catalyst 1 contributing through π–π interactions [5]. Based on Catalyst 1,further modifications to the axial ligand are anticipated to enhance catalytic performance.
Isoquinoline has a larger π-system than pyridine, and through stronger π-π interactions, it can enhance the rate and probability of O-O bond formation while lowering the energy barrier. Thus, replacing pyridine with isoquinoline results in Catalyst 2 (Figure 1). Kinetic measurements reveal that under pH=1 conditions, Catalyst 2 can achieve a TOF of 303 s−1, which is approximately 40 times higher than that of Catalyst 1 for Ce(IV)-driven water oxidation at pH 1. This strategy of enhancing catalytic efficiency by modifying axial ligands is a common approach [6].
Building on this, Yi et al. have replaced the axial ligands with negatively charged sulfonate groups and positively charged bipyridine, resulting in Catalyst 3. The axial ligands interact through electrostatic forces due to the attraction between positive and negative charges, increasing the probability of O-O bond formation. Kinetic measurements have revealed that Catalyst 3 exhibits a TOF of 12.4 s−1 under pH = 1 conditions, comparatively TOF was 7 s−1 for Catalyst 1 [7]. Subsequently, Tong et al. replaced the bipyridine in the Ru-based catalyst with phenanthroline, resulting in Catalyst 4. However, due to the increased hydrophobicity of phenanthroline, the face-to-face O-O bond formation pattern shifted to a back-to-back and face-to-back arrangement, significantly reducing the catalytic efficiency of the catalyst. Under the conditions of pH = 1, the TOF value is 0.04–0.1 s−1. Ce(IV)-driven water oxidation results and Density functional theory (DFT) calculations indicated that the O-O bond formation mechanism had shifted to water nucleophilic attack (WNA).
To enhance the catalytic efficiency of catalysts containing phenanthroline ligands, researchers continued to employ axial ligand modification strategies. To increase π-π interactions and improve the rate of O-O bond formation, the axial ligand was changed from pyridine to isoquinoline, resulting in Catalyst 5. This ligand modification approach can alter the O-O bond formation pattern from back-to-back and face-to-back to face-to-face, enhancing the catalytic efficiency of the catalyst, with a TOF value of 2-7 s−1 at pH = 1. Through DFT calculations, it was found that the O-O bond formation mechanism of Catalyst 5 is intermolecular interaction between two high-valent Ru-oxo species [8]. In addition to modifications of the axial ligands, Yang et al. have replaced the two carboxylic acid groups in the original Catalyst 1 with two sulfonic acid groups, resulting in Catalyst 6. Electrochemical measurements revealed that the sulfonic acid groups have a weaker electron-donating ability, which lowers the energy barrier for O-O bond formation. Under neutral conditions, the formation of a pentavalent ruthenium-oxo intermediate is not required; the O-O bond can be formed via an I2M mechanism among the tetravalent ruthenium-oxo intermediates. Catalyst 6 exhibits significantly higher electrocatalytic efficiency than Catalyst 1, with a TOF value of approximately 12900 s−1 in neutral conditions, albeit the driving forces are different [5].
To explore the catalytic efficiency of replacing only one carboxylic acid group in the original Catalyst 1, Yang et al. further designed Catalyst 7, which replaces one carboxylic acid group with a sulfonic acid group. Kinetic isotope effect studies and DFT calculations revealed that the O-O bond formation mechanism of Catalyst 7 differs from that of Catalyst 6 and does not proceed via a bimolecular mechanism. Under neutral conditions, Catalyst 7 can rapidly form the O-O bond between tetravalent ruthenium-oxo intermediates via the WNA mechanism. The TOF value is 1013 s−1 in neutral conditions, which is lower compared to the catalytic efficiency of Catalyst 6 but significantly higher than that of Catalyst 4, which shares the same O-O bond formation mechanism [9]. These catalysts represent the state-of-the-arts of the Ru-based bimolecular catalysts developed for water oxidation catalysis. The performance of the discussed catalysts, including their turnover frequencies (TOFs) and mechanisms, is summarized in Table 1.
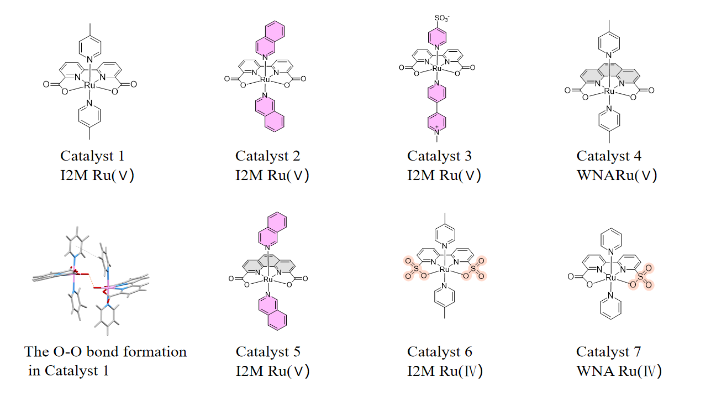
Figure 1: The molecular structure of the Ru catalysts discussed above.
Table 1: Comparison of the activity of the Ru catalysts (Figure 1) in water oxidation catalysis.
Catalyst | pH | TOFa | [CeⅣ]b | TOFc | Potential | Mechanism |
1 | 1 | 33 s-1 | 0.48 M | 7 s-1 | 1.8 V vs NHE | I2M |
2 | 1 | 303 s-1 | 0.48 M | / | / | I2M |
3 | 1 | 12.4 s-1 | 0.20 M | / | / | I2M |
4 | 1 | 0.04-0.1 s-1 | 0.17 M | / | / | WNA |
5 | 1 | 2-7 s-1 | 0.17 M | / | / | I2M |
6 | 7 | / | / | 12900 s-1 | 1.63 V vs NHE | I2M |
1 | / | / | 160 s-1 | 1.74 V vs NHE | I2M | |
7 | 7 | / | / | 1013 s-1 | 1.21 V vs NHE | WNA |
a. TOF for [CeⅣ]-driven water oxidation; b. Concentrations of CeⅣ and catalysts for [CeⅣ]-driven water oxidation; c. TOF for voltage-driven water oxidation.
2.2. Copper-based Catalysts
Due to the scarcity and high costs of noble metals, non-noble metal catalysts with equivalent activity, such as copper, iron, cobalt, and manganese, can be more ideal for catalysis. For copper, a binuclear copper strategy was proposed, investigating the O-O bond formation mechanism, and attempting to enhance catalytic efficiency by altering the type of ligands. In the case of Catalyst 1 in Figure 2, Su et al. employed a naphthyridine ligand, which is a bidentate ligand capable of coordinating with two copper ions simultaneously [10]. The synergistic effect of the dual active sites facilitates the removal of electrons from both copper atoms, preventing charge build-up. The ligand features a π-conjugated system that can modulate the electronic properties of the catalyst, ultimately preventing the formation of high oxidation state CuIV = O. DFT calculations confirmed that the O-O bond forms through intramolecular direct coupling between CuIII–O(H) and a μ-oxo without the need for intermediate CuIV = O formation. The formation pathway of the O-O bond is depicted in Figure 3. The TOF value of Catalyst 1 under neutral conditions is 0.6 s-1.
Since peptide-like compounds can stabilize high oxidation state metal ions, Ruan et al. have designed a binuclear copper-peptide-like complex, which utilizes a peptide-like trimer as Catalyst 2 in Figure 2 [11]. In this complex, the bipyridine ligand, with its π-conjugated system, can modulate electronic properties and optimize redox properties, thereby enhancing catalytic activity. The two ethanol groups act as proton relays in Proton-coupled Electron Transfer (PCET) and stabilize intermediates through hydrogen bonding, reducing the reaction energy barrier. Unlike other studies, Catalyst 2 exhibits stable and rapid performance in borate buffer solution. This is attributed to the borate solution acting as an oxygen source, facilitating its participation in the O-O bond formation. DFT calculations also reveal that the O-O bond formation mechanism is intramolecular oxygen-oxygen coupling between CuIII−O• and borate. The formation pathway of the O-O bond is depicted in Figure 3. Electrochemical measurements yield a TOF value of 129 s−1 at pH = 9.35.
Apart from binuclear copper, Jiang et al. have also designed a tetranuclear copper catalyst, namely Catalyst 3 (Figure 2) [12]. This catalyst utilizes Schiff base ligands with multiple coordination sites, which can tightly connect metal centers to form polymetallic clusters, creating a complex with a synergistic effect of a tetranuclear copper center. On this basis, glycine and glutamic acid are chosen to coordinate with copper ions. Electrochemical measurements show that the complex with glycine coordination has a TOF value of 267 s−1 at pH = 12, whilst that achieved by the complex with glutamic acid coordination was about 105 s−1 under the same condition. The activity difference between the two catalysts lies in the dissociated state, the carboxylate anion can act as a ligand to form additional coordination bonds with the copper center and alter the geometric structure of the copper center, enhancing the binding capacity to the substrate and catalytic activity. In addition to the issue of ligand types, researchers are also exploring the symmetry of ligands within the catalyst molecules. Hu et al. have designed catalyst 1 and Catalyst 4 (Figure 2) [13]. Both catalysts can avoid the formation of high-valent CuIV=O and form O-O bonds through a binuclear cooperative pathway. Catalyst 1 is symmetrical, assisting in O-O bond formation with a semi-chelated pyridyl group. Catalyst 4 is asymmetrical, lacking one pyridyl group compared to Catalyst 1 and has an open coordination site, allowing water to participate in the catalysis more readily. It also exhibits a significant kinetic isotope effect, which can optimize the proton transfer step. The asymmetrical catalyst lacks an intramolecular proton acceptor, and the buffer solution can assist in proton transfer.
In addition to ligand modifications, researchers are also dedicated to studying the impact of linker arms on catalyst activity. Emphasizing the investigation of the geometric structure of catalysts on O-O bond formation, Chen et al. have designed three binuclear complexes with different linker arms to delve into the relationship between O-O bond formation and optimal bonding geometry [14]. The linkers in Catalysts 7, 1, and 8 (Figure 2) are Biologically Inspired Processing and Analysis of Neural Modules (BPMAN) Cu2, 6-hydroxypicolinic acid (6-HPA) Cu2, and bpy-6,6'-dimethylamide (BPMAD) Cu2, respectively, with rigidity order as Catalyst 7 > Catalyst 1 > Catalyst 8. At pH = 7, the TOF values correspond to the rigidity order, being 2.01 s−1, 0.6 s−1, and 0.17 s−1, respectively. Thus, it is concluded that catalytic activity correlates positively with linker arm rigidity, with O-O bond formation occurring via intramolecular oxygen-oxygen coupling. Apart from rigid linkers, Zhang et al. designed a binuclear copper complex with a flexible linker arm, namely Catalyst 5 (Figure 2) [15]. The introduction of a flexible linker arm allows free rotation and positional changes of the two active centers, promoting cooperative action. DFT calculations reveal that flexible linker arms can lower the catalytic reaction energy barrier from 21.7 kcal/mol to 9.1 kcal/mol. Additionally, the linker arm contains pyridine, providing a π-conjugated system that is beneficial for electron transfer. The O-O bond formation follows a single-molecule dual-site mechanism (Figure 3), with a TOF value of 144 s−1 at pH = 12 conditions. For a direct comparison, the authors also designed a monomeric counterpart, Catalyst 6 (Figure 2). DFT calculations indicate that the catalytic process of this monomeric counterpart requires the formation of high-valent copper-oxygen intermediates with a higher energy barrier, and the O-O bond formation mechanism is the WNA mechanism, with a TOF value of 4.86 s−1 under pH = 12 conditions. To provide a clear comparative overview of the catalytic efficiencies and mechanisms discussed for each catalyst, the key performance metrics, including turnover frequency (TOF), pH conditions, and mechanistic pathways, are summarized in Table 2.
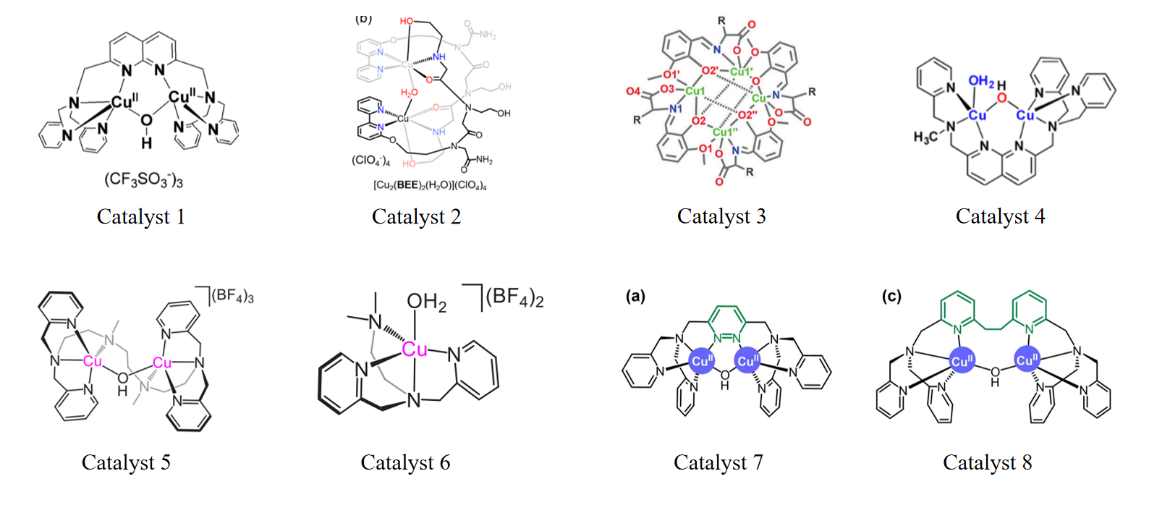
Figure 2: The molecular structure of the Cu catalysts discussed above.
Table 2: Comparison of the activity of the Cu catalysts (Figure 2) in water oxidation catalysis.
Catalyst | pH | Potential | TOF | Mechanism |
1 | 7 | 1.87 V vs NHE | 0.6 s-1 | 1 |
2 | 9.35 | 1.8 V vs NHE | 129 s-1 (CV)5503 s-1 (FOWA) | 2 |
3 (Gly) | 12 | 1.7 V vs NHE | 267 s-1 | 3 |
3 (Glu) | 12 | 1.56 V vs NHE | 105 s-1 | 3 |
4 | 7 | 1.87 V vs NHE | 0.78 s-1 | 1 |
5 | 12 | 1.5 V vs NHE | 144 s-1 | 4 |
6 | 12 | 1.5 V vs NHE | 4.86 s-1 | 5 |
7 | 7 | 1.87 V vs NHE | 0.17 s-1 | 1 |
8 | 7 | 1.87 V vs NHE | 2.01 s-1 | 1 |
In the table, the mechanism for O-O bond formation, Mechanism 1, 2, 4 are depicted in Figure 3; Mechanism 3 corresponds to I2M; Mechanism 5 is referred to as WNA.
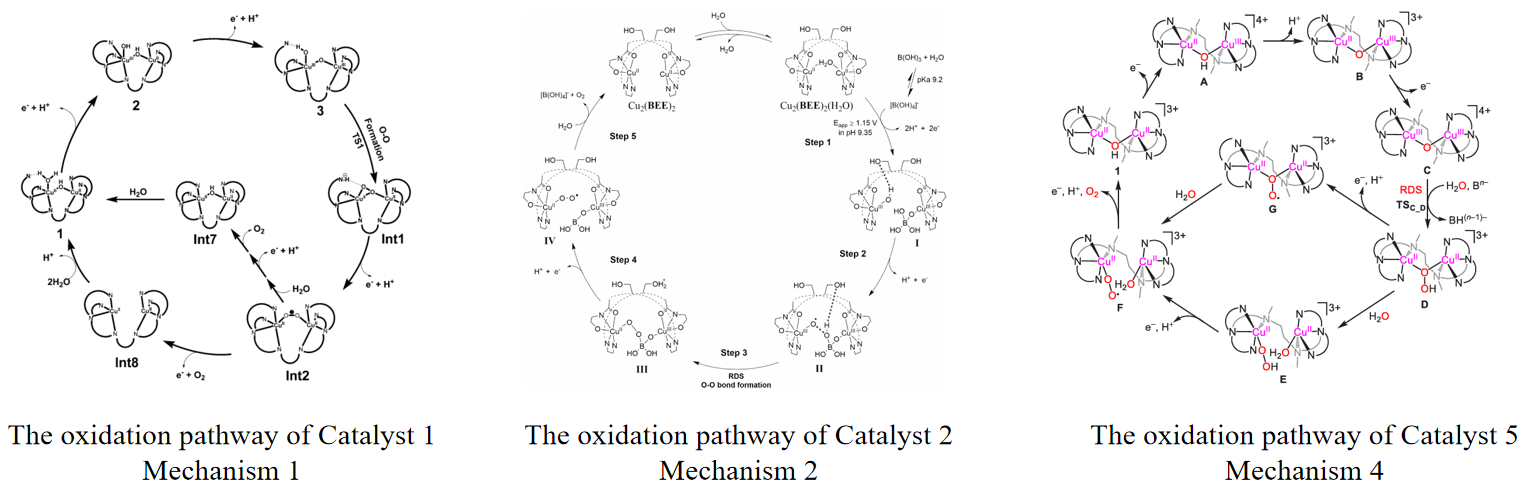
Figure 3: The oxidation pathway of Catalyst 1, 2, 5.
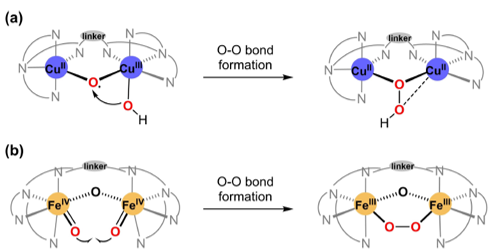
Figure 4: Divergent O-O bond formation modes in Cu-Core and Fe-Core catalysts.
2.3. Ferrum-based Catalysts
Due to the abundant presence of Fe in nature, iron-core catalysts also hold significant research value. It can be observed that the O-O bond formation in Cu catalysts 7 and 8 (Figure 2) involves the oxygen on the metal and the oxygen in the oxo-bridge. In contrast, Figure 4 illustrates that in iron-core catalysts, the bond is formed between the oxygens on the two metals, indicating a distinct mechanistic difference. Below, the paper analyzes the mechanism of the iron core catalyst.
Wickramasinghe et al. synthesized a binuclear iron complex FeIII(ppq) containing a novel tetradentate ligand ppq (2-(pyrid-2″-yl)-8(1″,10″-phenanthrolin-2″-yl)-quinoline), which is referred to as Catalyst 4 in Figure 5 [16]. The ligand features a tetrapyridine framework with an additional sp²-hybridized center at the quinoline C8 position, providing a square planar coordination environment for the iron center. Over a period of more than 6 hours of observation, there were no changes in the UV-vis spectra of Catalyst 4, indicating that the dimeric catalyst remains intact in aqueous solution. The oxygen evolution rate of Catalyst 4 follows first-order kinetics with respect to the catalyst concentration in presence of CeIV, with a TOF of 7920 h⁻¹. Subsequently, the reaction solution after the cessation of oxygen generation was examined using dynamic light scattering, and no formation of iron oxide nanoparticles was detected, suggesting that the catalyst remains stable during the turnover. Electrochemical results suggest that the FeIII(ppq) complex undergoes a two-electron oxidation followed by a disproportionation process, wherein the FeIIIFeV = O intermediate acts as the active species in water oxidation process, enabling the further oxidation of water molecules to generate oxygen.
Further research into iron-core catalysts has focused on ligand modifications for enhanced efficiency. Zhang et al. developed three dinuclear iron complexes by adjusting ligand rigidity, namely [Fe2(μ-O)(OH2)2(TPA)2]4+ (1), [Fe2(μ-O)(OH2)2(6-HPA)]4+ (2), and [Fe2(μ-O)(OH2)2(BPMAN)]4+ (3), as electrocatalysts in alkaline conditions (0.1 M NaHCO3, pH 8.4) [17]. Complexes 1 and 2 effectively catalyzed water oxidation via oxo–oxo coupling, while the rigidity in complex 3’s BPMAN ligand hindered isomerization necessary for O-O bond formation, diminishing its catalytic activity. DFT calculations highlighted ligand flexibility as crucial for this process. These results elucidate that the O-O bond is formed through intramolecular oxo/oxo or oxo/oxyl coupling, with bimetallic synergism and ligand rigidity profoundly affecting catalytic performance, offering insights for designing iron-based catalysts.
Li et al. proposed an alternative reaction mechanism based on DFT results. The Catalyst 4 undergoes two PCET processes to generate intermediate [Cl–FeIV–O–FeIV=O]3+ [18]. The first PCET process occurs at 1.66 V to form [Cl–FeIII–O–FeIV–OH]3+, and the second PCET process requires 1.44 V to produce complex [Cl–FeIV–O–FeIV=O]3+. Subsequent calculation revealed that further oxidation to [Cl–FeIV–O–FeV=O]4+ necessitates a potential of 2.15 V, which cannot be achieved under experimental conditions, thus avoiding reliance on further high-potential oxidation steps. The article also proposes a mechanism for O-O bond formation, where complex [Cl–FeIV–O–FeIV=O]3+ is nucleophilically attacked by chloride ions, breaking down into two subunits to initiate O-O bond formation, which is coupling of two [FeIV=O]+ radicals.
Masaoka et al. described a pentanuclear iron-based water oxidation catalyst with neighboring coordinatively unsaturated active sites to facilitate water binding, activation, and O–O bond formation, referring to Catalyst 6, which is depicted in Figure 6 [19]. In the absence of water, the CV exhibited five reversible redox waves at −0.55, 0.13, 0.30, 0.68, and 1.08 V versus Fc/Fc⁺ (ferrocene/ferrocenium couple), corresponding to sequential one-electron oxidations of the iron centers from FeII₅ to FeIII₅. Adding about 10% water led to a significant catalytic current after the fourth redox peak, with TOF values from 140 to 1400 s−1. Electrochemical, UV-vis, and X-ray Photoelectron Spectroscopy analyses confirmed the catalyst's homogeneous nature. The catalytic mechanism was proposed based on crystal structures of reaction intermediates, electrochemical and spectroelectrochemical measurements, 57Fe Mössbauer spectroscopy, and DFT calculations. The catalyst first oxidized to FeIII₅ species (S4) and then reacted with water to form FeIII₅(OH2) state (A) with an estimated reaction barrier of 15 kcal/mol. Coordination with another water molecule, coupled with four proton transfers, produced a key reaction intermediate (B) containing two FeIV–oxo sites. DFT calculations suggested that electron rearrangement among the five iron centers generated two co-facial FeIV = O sites, where the O–O bond formed intramolecularly with an estimated reaction barrier of less than 10 kcal/mol. The catalyst mimics the mechanism of the Oxygen Evolving Center (OEC) by accumulating multiple charges on multinuclear sites, demonstrating that multi-site assisted charge transfer and dual-site O–O bond formation represent a promising strategy for catalyst design.
Incorporating other metals into iron-core catalysts to form heterometallic catalysts is a novel exploratory approach. Inspired by the heterometallic metal catalyst CaMn4O5 in OEC, the authors designed a heterometallic catalyst with Ni and Fe, referred to as Catalyst 5 (Figure 5) [20]. Through the analysis of structure-activity relationships with ZnII ion-substituted heterometallic binuclear complexes, it was determined that Ni and Fe are both indispensable in the catalysis jointly participated by the NiFe center. The O-O bond formation mechanism of this complex involves a series of oxidations to form a high-valent NiIII(μ-O)FeIV core, in which the bridging O atom and the terminal FeIV = O group form the O-O bond through oxyl-oxo coupling. In addition, the authors confirmed that FeFe and NiNi analogs are not suitable as water oxidation catalysts. The FeFe complex remains stable but exhibits no activity, whereas the NiNi catalyst is readily converted into active NixOy nanoparticles during the electrocatalysis process. In contrast, the NiFe catalyst remains stable in long-term electrolysis, as confirmed by UV-Vis spectral tests. The catalyst exhibits a TOF value of 11.5 s−1 at pH = 10 and applied potential of 1.4 V vs NHE, as summarized in Table 3, which compares the activity of the Fe catalysts.
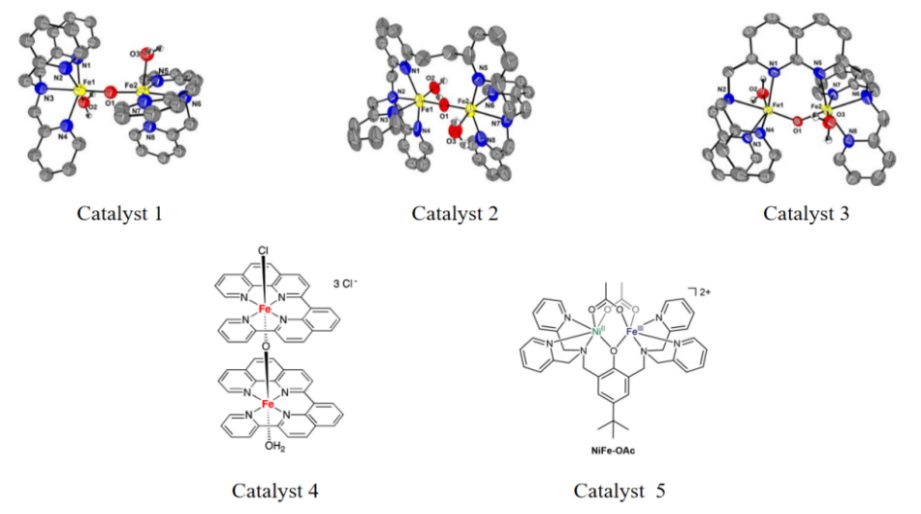
Figure 5: The molecular structure of the Fe catalysts discussed above.
Table 3: Comparison of the activity of the Fe catalysts.
Catalyst | pH | TOF | Potential | Ce |
1 | 8.4 | 0.55 s-1 | / | / |
2 | 8.4 | 0.04 s-1 | / | / |
3 | 8.4 | / | / | / |
4 | 1 | 7920 h-1 | / | 0.1 M |
5 | 10 | 11.5 s-1 | 1.26 V vs NHE | / |
6 | 7 | 1900 s-1 | 1.8 V vs Fc/Fc+ | / |
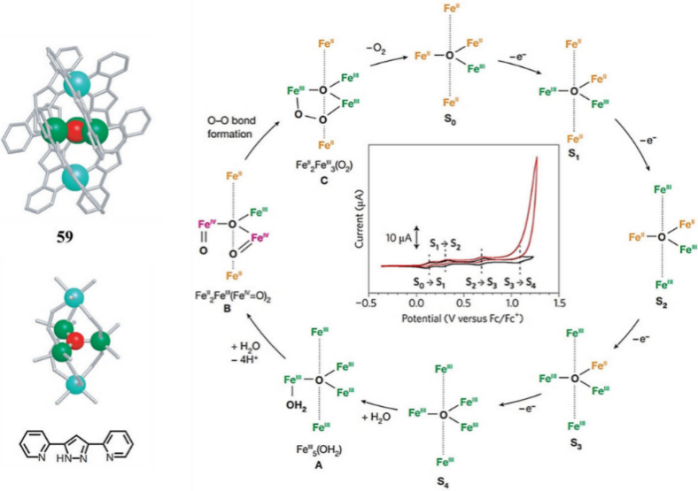
Figure 6: The molecular structure and proposed mechanism for pentanuclear iron-based electrocatalyst (Catalyst 6).
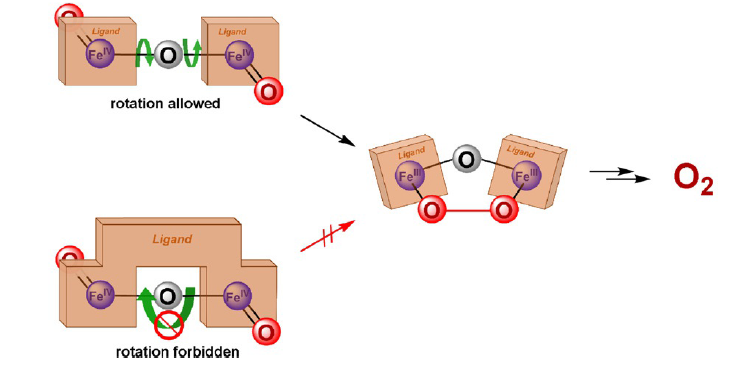
Figure 7: The O-O bond formation of Catalyst 1, Catalyst 2, and Catalyst 3.
2.4. Cobalt-based Catalysts
Apart from Cu and Fe, Co has also been utilized in water oxidation catalysts. Here, only one is cited as a representative example. Xie et al. designed a binuclear hydrazide-bridged ligand (LH4) with penta- and hexa-coordinate sites, and synthesized the binuclear cobalt-based catalyst [(L4−)CoIII2(OH)] by reacting the LH4 with Co(ClO4)2·6H2O in methanol in Figure 8 [21]. This novel catalyst achieves electrocatalytic water oxidation under pH 7 with a TOF of 2.2 s−1 at an applied potential 1.8 V vs NHE. Regarding the formation pathway of the O-O bond, the catalyst [(L4−)CoIII2(OH)]+ is first oxidized through a PCET process to generate a mixed-valence intermediate [CoIIICoIV=O]+. The generated intermediate is further oxidized to form the [CoIVCoIV=O]2+ intermediate. The [CoIVCoIV=O]2+ intermediate reacts with water to form the O-O bond via a WNA mechanism, producing the [CoIIICoIII-OOH]+ intermediate, with the electrons from the attacking water molecule being distributed across both Co centers.
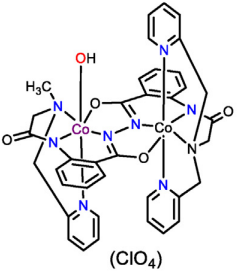
Figure 8: The structure of [(L4-)Co2(OH)](ClO4).
2.5. Manganese-based Catalysts
Aside from Cobalt, Manganese has also been employed in water oxidation catalysis. Given that the OEC is a manganese-oxo cluster, researchers have mimicked this structure in the design of the catalyst [Mn12O12(O2CC6H2(OH)3)16(H2O)4] (Mn12TH) [22]. Previous Mn-based nuclear catalysts were not highly efficient, prompting the researchers in this study to modify the ligand from 2,3-dihydroxybenzoic acid (dhbH) to 3,4,5-trihydroxybenzoic acid (thbH). Electrocatalytic activity measurements revealed that this minor alteration led to an almost three orders of magnitude faster (kobs = 22 s-1) and more efficient (93% FE) catalytic water oxidation activity, with an overpotential of only 74 mV at pH 6, which is the lowest overpotential reported to date for homogeneous water oxidation electrocatalysts. The authors propose that the hydroxyl groups on the ligand may facilitate catalysis by promoting the formation of high-valent species required for water oxidation through ligand-assisted PCET as shown in Figure 9. Electron transfer occurs not only on the Mn center, but also involves the non-innocent phenolate ligand, which gets oxidized to a quinone during the catalytic process, accompanied by the change in the para-carboxylate anion (from -C(OH)=O to =C(OH)2). The latter is more favorable for stabilizing the high-valent Mn state.
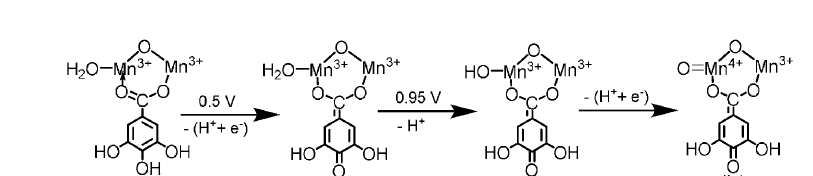
Figure 9: The possible oxidation pathway of Mn center in 0.1 M acetate buffer solution at pH 6.
3. Conclusion
This paper presents a comprehensive review on the design strategies for dual-site catalysts based on different metals, especially transition ones, which include the modification of lateral and axial ligands, doping with heterogenous elements, and the design of hetero-nuclear and multi-nuclear metals. The roles of dual-site encompass: the formation of O-O bonds via the I2M pathway, which circumvents the formation of high-energy intermediates such as M-OOH through the WNA pathway; electron transfer occurs at the dual-sites, preventing the accumulation of charge at single sites; through multi-site synergy, it reveals other O-O bond formation methods beyond WNA and I2M, thereby enhancing the understanding of natural photosynthesis, where catalytic centers comprise multi-nuclear structures whose mechanisms remain debated.
Based on summary of the relevant state-of-the-arts, the dual-site homogeneous water oxidation catalysts (molecular catalysts), and the dual-site strategy could be extended to heterogeneous water oxidation catalysts. Introducing Mn atoms into the RuO2 catalyst to create Mn4-δ-O-Ru4+δ dual-active sites with local structural symmetry but oxidation state asymmetry can transform the traditional adsorbate evolution mechanism into the oxide path mechanism, significantly enhancing efficiency [23]. Similarly, the A-Fe2S1N5/SNC catalyst enhances catalytic performance by configuring asymmetric iron diatomic sites (Fe-Fe) on highly defective nitrogen-sulfur co-doped carbon nanosheets (SNC) [24]. In another example, Ni-Fe dual-site catalysts, utilizing graphitic carbon nitride as a support, demonstrate substantial performance improvements in heterogeneous catalysis for the oxygen evolution reaction [25]. Heterogeneous catalysts possess a higher practical value in industry. Broadly speaking, the dual-site strategy can also be extended to other energy-related catalytic applications, such as oxygen reduction (the reverse reaction of water oxidation): by precisely regulating the electronic structure and spin state of metal sites, dual-metal atomically dispersed Fe,Mn/N-C catalysts have been designed to achieve high activity and stability of Oxygen Reduction Reaction catalysts in both alkaline and acidic media [26]. Moreover, dual-site catalysts incorporating platinum-group metal atoms have been designed to enhance the performance of copper-based catalysts in CO2 reduction, particularly in converting CO2 into energy-dense hydrocarbon molecules, such as CH4 and C2H4 [27].
Overall, these advancements underscore the versatility and broad applicability of dual-site catalysts in enhancing the efficiency of various catalytic processes essential for energy conversion and environmental sustainability.
References
[1]. Shivanna, K.R. (2022) Climate change and its impact on biodiversity and human welfare. Indian Natl. Sci. Acad., 88(2), 160-171.
[2]. Tashie-Lewis, B.C. and Nnabuife, S.G. (2021) Hydrogen Production, Distribution, Storage and Power Conversion in a Hydrogen Economy - A Technology Review. Chem. Eng. J. Adv., 8, 100172.
[3]. Hassan, Q., Algburi, S., Sameen, A.Z., Salman, H.M. and Jaszczur, M. (2024) Green hydrogen: A pathway to a sustainable energy future. Int. J. Hydrogen Energy, 50, 310-333.
[4]. Paygozar, S., Aghdam, A.S.R., Hassanizadeh, E., Andaveh, R. and Darband, G.B. (2023) Recent progress in non-noble metal-based electrocatalysts for urea-assisted electrochemical hydrogen production. Int. J. Hydrogen Energy, 48(20), 7219-7259.
[5]. Yang, J. et al. (2021) From Ru-bda to Ru-bds: a step forward to highly efficient molecular water oxidation electrocatalysts under acidic and neutral conditions. Nat. Commun., 12, 373.
[6]. Duan, L. et al. (2012) A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nature Chem., 4, 418–423.
[7]. Yi, J. et al. (2021) Electrostatic Interactions Accelerating Water Oxidation Catalysis via Intercatalyst O–O Coupling. J. Am. Chem. Soc., 143(6), 2484-2490.
[8]. Liu, T. et al. (2024) Intermolecular O–O Bond Formation between High-Valent Ru–oxo Species. Inorg. Chem., 63(35), 16161-16166.
[9]. Yang, J. et al. (2024) Adaptive water oxidation catalysis on a carboxylate-sulfonate ligand with low onset potential. Chem. Commun., 60(48), 6162-6165.
[10]. Su, X.J. et al. (2015) Electrocatalytic Water Oxidation by a Dinuclear Copper Complex in a Neutral Aqueous Solution. Angew. Chem., 54(16), 4909-4914.
[11]. Ruan, G., Ghosh, P., Fridman, N. and Maayan, G. (2021) A Di-Copper-Peptoid in a Noninnocent Borate Buffer as a Fast Electrocatalyst for Homogeneous Water Oxidation with Low Overpotential. J. Am. Chem. Soc., 143(28), 10614-10623.
[12]. Jiang, X. et al. (2018) A Bio-inspired Cu4O4 Cubane: Effective Molecular Catalysts for Electrocatalytic Water Oxidation in Aqueous Solution. Angew. Chem., 130(26), 7976-7980.
[13]. Hu, Q.Q., Su, X.J. and Zhang, M.T. (2018) Electrocatalytic Water Oxidation by an Unsymmetrical Di-Copper Complex. Inorg. Chem., 57(17), 10481-10484.
[14]. Chen, Q.F., Xian, K.L., Zhang, H.T., Su, X.J., Liao, R.Z. and Zhang, M.T. (2024) Pivotal Role of Geometry Regulation on O−O Bond Formation Mechanism of Bimetallic Water Oxidation Catalysts. Angew. Chem., 63(9).
[15]. Zhang, X., Li, Y.Y., Jiang, J., Zhang, R., Liao, R.Z. and Wang, M. (2020) A Dinuclear Copper Complex Featuring a Flexible Linker as Water Oxidation Catalyst with an Activity Far Superior to Its Mononuclear Counterpart. Inorg. Chem., 59(8), 5424-5432.
[16]. Wickramasinghe, L.D., Zhou, R., Zong, R., Vo, P., Gagnon, K.J. and Thummel, R.P. (2015) Iron Complexes of Square Planar Tetradentate Polypyridyl-Type Ligands as Catalysts for Water Oxidation. J. Am. Chem. Soc., 137(41), 13260–13263.
[17]. Zhang, H.T., Su, X.J., Liao, R.Z. and Zhang, M.T. (2021) Iron-Catalyzed Water Oxidation: O–O Bond Formation via Intramolecular Oxo–Oxo Interaction. Angew. Chem., 60(22), 12467-12474.
[18]. Li, G. and Ahlquist, M.S.G. (2024) O–O bond formation via radical coupling in a dinuclear iron water oxidation catalyst with high catalytic activity. Dalton Trans., 53, 2456-2459.
[19]. Okamura, M. et al. A pentanuclear iron catalyst designed for water oxidation. Nature, 530, 465–468.
[20]. Zhang, H.T., Guo, Y.H., Xiao, Y., Du, H.Y. and Zhang, M.T. (2023) Heterobimetallic NiFe Cooperative Molecular Water Oxidation Catalyst. Angew. Chem., 62(18), e202218859.
[21]. Xie, F. and Zhang, M.T. (2021) Bimetallic water oxidation: One-site catalysis with two-sites oxidation. J. Energy Chem., 63, 1-7.
[22]. Ghosh, T. and Maayan, G. (2019) Efficient Homogeneous Electrocatalytic Water Oxidation by a Manganese Cluster with an Overpotential of Only 74 mV. Angew. Chem., 58(9), 2785-2790.
[23]. Ji, Q. et al. (2024) Operando identification of the oxide path mechanism with different dual-active sites for acidic water oxidation. Nat. Commun., 15, 8089.
[24]. Zhang, L. et al. (2024) High-density asymmetric iron dual-atom sites for efficient and stable electrochemical water oxidation. Nat. Commun., 15, 9440.
[25]. Wan, W. et al. (2021) Mechanistic insight into the active centers of single/dual-atom Ni/Fe-based oxygen electrocatalysts. Nat. Commun., 12, 5589.
[26]. Yang, G. et al. (2021) Regulating Fe-spin state by atomically dispersed Mn-N in Fe-N-C catalysts with high oxygen reduction activity. Nat. Commun., 12, 1734.
[27]. Chhetri, M. et al. (2023) Dual-site catalysts featuring platinum-group-metal atoms on copper shapes boost hydrocarbon formations in electrocatalytic CO2 reduction. Nat. Commun., 14, 3075.
Cite this article
Huang,Y. (2025). Molecular Water Oxidation Catalysts with Dual-active Sites. Applied and Computational Engineering,129,162-173.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 5th International Conference on Materials Chemistry and Environmental Engineering
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Shivanna, K.R. (2022) Climate change and its impact on biodiversity and human welfare. Indian Natl. Sci. Acad., 88(2), 160-171.
[2]. Tashie-Lewis, B.C. and Nnabuife, S.G. (2021) Hydrogen Production, Distribution, Storage and Power Conversion in a Hydrogen Economy - A Technology Review. Chem. Eng. J. Adv., 8, 100172.
[3]. Hassan, Q., Algburi, S., Sameen, A.Z., Salman, H.M. and Jaszczur, M. (2024) Green hydrogen: A pathway to a sustainable energy future. Int. J. Hydrogen Energy, 50, 310-333.
[4]. Paygozar, S., Aghdam, A.S.R., Hassanizadeh, E., Andaveh, R. and Darband, G.B. (2023) Recent progress in non-noble metal-based electrocatalysts for urea-assisted electrochemical hydrogen production. Int. J. Hydrogen Energy, 48(20), 7219-7259.
[5]. Yang, J. et al. (2021) From Ru-bda to Ru-bds: a step forward to highly efficient molecular water oxidation electrocatalysts under acidic and neutral conditions. Nat. Commun., 12, 373.
[6]. Duan, L. et al. (2012) A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nature Chem., 4, 418–423.
[7]. Yi, J. et al. (2021) Electrostatic Interactions Accelerating Water Oxidation Catalysis via Intercatalyst O–O Coupling. J. Am. Chem. Soc., 143(6), 2484-2490.
[8]. Liu, T. et al. (2024) Intermolecular O–O Bond Formation between High-Valent Ru–oxo Species. Inorg. Chem., 63(35), 16161-16166.
[9]. Yang, J. et al. (2024) Adaptive water oxidation catalysis on a carboxylate-sulfonate ligand with low onset potential. Chem. Commun., 60(48), 6162-6165.
[10]. Su, X.J. et al. (2015) Electrocatalytic Water Oxidation by a Dinuclear Copper Complex in a Neutral Aqueous Solution. Angew. Chem., 54(16), 4909-4914.
[11]. Ruan, G., Ghosh, P., Fridman, N. and Maayan, G. (2021) A Di-Copper-Peptoid in a Noninnocent Borate Buffer as a Fast Electrocatalyst for Homogeneous Water Oxidation with Low Overpotential. J. Am. Chem. Soc., 143(28), 10614-10623.
[12]. Jiang, X. et al. (2018) A Bio-inspired Cu4O4 Cubane: Effective Molecular Catalysts for Electrocatalytic Water Oxidation in Aqueous Solution. Angew. Chem., 130(26), 7976-7980.
[13]. Hu, Q.Q., Su, X.J. and Zhang, M.T. (2018) Electrocatalytic Water Oxidation by an Unsymmetrical Di-Copper Complex. Inorg. Chem., 57(17), 10481-10484.
[14]. Chen, Q.F., Xian, K.L., Zhang, H.T., Su, X.J., Liao, R.Z. and Zhang, M.T. (2024) Pivotal Role of Geometry Regulation on O−O Bond Formation Mechanism of Bimetallic Water Oxidation Catalysts. Angew. Chem., 63(9).
[15]. Zhang, X., Li, Y.Y., Jiang, J., Zhang, R., Liao, R.Z. and Wang, M. (2020) A Dinuclear Copper Complex Featuring a Flexible Linker as Water Oxidation Catalyst with an Activity Far Superior to Its Mononuclear Counterpart. Inorg. Chem., 59(8), 5424-5432.
[16]. Wickramasinghe, L.D., Zhou, R., Zong, R., Vo, P., Gagnon, K.J. and Thummel, R.P. (2015) Iron Complexes of Square Planar Tetradentate Polypyridyl-Type Ligands as Catalysts for Water Oxidation. J. Am. Chem. Soc., 137(41), 13260–13263.
[17]. Zhang, H.T., Su, X.J., Liao, R.Z. and Zhang, M.T. (2021) Iron-Catalyzed Water Oxidation: O–O Bond Formation via Intramolecular Oxo–Oxo Interaction. Angew. Chem., 60(22), 12467-12474.
[18]. Li, G. and Ahlquist, M.S.G. (2024) O–O bond formation via radical coupling in a dinuclear iron water oxidation catalyst with high catalytic activity. Dalton Trans., 53, 2456-2459.
[19]. Okamura, M. et al. A pentanuclear iron catalyst designed for water oxidation. Nature, 530, 465–468.
[20]. Zhang, H.T., Guo, Y.H., Xiao, Y., Du, H.Y. and Zhang, M.T. (2023) Heterobimetallic NiFe Cooperative Molecular Water Oxidation Catalyst. Angew. Chem., 62(18), e202218859.
[21]. Xie, F. and Zhang, M.T. (2021) Bimetallic water oxidation: One-site catalysis with two-sites oxidation. J. Energy Chem., 63, 1-7.
[22]. Ghosh, T. and Maayan, G. (2019) Efficient Homogeneous Electrocatalytic Water Oxidation by a Manganese Cluster with an Overpotential of Only 74 mV. Angew. Chem., 58(9), 2785-2790.
[23]. Ji, Q. et al. (2024) Operando identification of the oxide path mechanism with different dual-active sites for acidic water oxidation. Nat. Commun., 15, 8089.
[24]. Zhang, L. et al. (2024) High-density asymmetric iron dual-atom sites for efficient and stable electrochemical water oxidation. Nat. Commun., 15, 9440.
[25]. Wan, W. et al. (2021) Mechanistic insight into the active centers of single/dual-atom Ni/Fe-based oxygen electrocatalysts. Nat. Commun., 12, 5589.
[26]. Yang, G. et al. (2021) Regulating Fe-spin state by atomically dispersed Mn-N in Fe-N-C catalysts with high oxygen reduction activity. Nat. Commun., 12, 1734.
[27]. Chhetri, M. et al. (2023) Dual-site catalysts featuring platinum-group-metal atoms on copper shapes boost hydrocarbon formations in electrocatalytic CO2 reduction. Nat. Commun., 14, 3075.