







1. Introduction
Prodrug is a chemical compound that has no activity or weak activity by itself, but after transformation in living organisms, it produces metabolites or original drugs with pharmacological activity or significantly enhanced pharmacological activity. The concept of a prodrug was first proposed by Adrien Albert in 1958, who defines a biologically active precursor, that is, a derivative form of a drug molecule which is not pharmacologically active in itself, but can be converted into a parent drug with pharmacological effects through enzymatic reactions or chemical transformations in vivo [1]. This strategy aims to optimize the ADMET (absorption, distribution, metabolism, excretion, and toxicity) characteristics of drugs through the design of prodrugs to improve the efficacy and safety of drugs. The release mechanism of a prodrug can be classified into three types: before, during, or after absorption, the active drug is released from its inactive form to achieve precise action on the target site. Some prodrug designs allow the active drug to be released only after it reaches a specific target of action, increasing the targeting and efficacy of the drug.
Prodrugs fall into two broad categories. One is the carrier-linked prodrug, which are conjugates of an active pharmaceutical agent with a carrier moiety. These conjugates exhibit diminished or negligible in vitro activity, yet undergo biotransformation in vivo through enzymatic or non-enzymatic pathways, culminating in the liberation of the parent drug and the manifestation of its therapeutic effects. Optimally, the carrier should possess attributes such as non-immunogenicity, facile synthesis, cost-effectiveness, stability under prodrug administration conditions, and biodegradability into inert metabolites [2]. The other is biological prodrug, which are novel entities derived from the molecular modification of bioactive compounds. These entities serve as substrates for metabolic enzymes, with the resultant enzymatic metabolites representing the anticipated bioactive species. With the continuous deepening of biomedical research and the rapid development of technical means, prodrugs are becoming more complex, from small molecule drugs [3] to now their application scope has been expanded to nucleic acid prodrugs [4], protein prodrugs [5], nanoprodrugs [6] and antibody prodrugs [7], providing new ideas and tools for the treatment of cancer, viral diseases, neurodegenerative diseases and other refractory diseases, but at the same time increasing the difficulty of design. For example, in the field of cancer treatment, specific pH, glutathione [8], ROS [9] and some specific enzymes in cancer cells provide more ideas for the design of prodrugs targeting tumor cells, especially the design of nano-prodrugs for the treatment of cancer [10] has become a hot topic. In addition, the study of intelligent prodrugs and activation mechanisms, such as photosensitive prodrugs [11] and enzyme-sensitive prodrugs, has opened a new path for precise control of the time, space and intensity of drug action.
This review article delineates an extensive array of prodrug classes, encompassing the phosphate prodrugs, ketone prodrugs, ester prodrugs, amide prodrugs, pH-responsive prodrugs, enzyme-activated prodrugs, carbamate prodrugs, liposomal prodrugs, and pleiotropic prodrugs. The collective utility of prodrugs is underscored by their multifaceted benefits, which include the amelioration of drug pharmacokinetics, the refinement of targeting precision, the attenuation of toxic effects, the amelioration of solubility profiles, the enhancement of drug payload capacity, and the optimization of bioavailability. The discussion within this review further elaborates on the prevalent chemical strategies for prodrug modification and their respective applications, highlighting the significance of these methodologies in advancing drug delivery and therapeutic efficacy.
1.1. Phosphate Prodrug
Phosphate prodrug is a special drug design strategy whose main purpose is to improve the pharmacokinetic properties of the drug, enhance the targeting, stability, and bioavailability of the drug, while reducing the toxic side effects of the drug, and can mask the unpleasant odor of the drug, improve water solubility, and thus improve the route of administration [12]. Phosphate prodrugs can be hydrolyzed in the body by the action of enzymes to release the active drug by linking the hydroxyl group on the active molecule with the phosphate ester group. ProTide technology [13] is a phosphoprodrug method that effectively improves the efficiency of intracellular drug delivery by partially masking the hydroxyl groups of nucleoside analogues monophosphates and monophosphonates with aromatic groups and amino acid esters, which are enzymatically cleaved intracellularly, releasing free nucleoside monophosphates and monophosphonates. This technology has been widely used in drug discovery and has led to the discovery of two FDA-approved (antiviral) ProTides. The advantages of phosphate ester prodrugs include improvement in pharmacokinetic parameters such as terminal elimination half-life, maximum plasma concentration, clearance, oral bioavailability, etc., showing promising trends. Modulating lipophilicity is a critical part of prodrug success, which is important for drug development regardless of clinical application. Additionally, phosphorylation is one of the important methods for the design of prodrugs containing hydroxyl drugs, which can be classified according to the valence state of the central phosphorus atom and the structure of the compound, and synthesized using different phosphoesterification reagents, which show their own advantages and limitations in the application of drugs. Through structural modification and optimization, phosphate ester prodrugs have been applied and improved in terms of physical and chemical properties (fat-soluble and water-soluble), drug safety, and pharmacokinetic properties [14].
One of the examples is Gemcitabine (GEM). GEM is an antimetabolite used in the treatment of a variety of cancers [15]. Its clinical application is limited by rapid metabolic inactivation, low oral bioavailability, and potential drug resistance. To address these issues, the investigators have developed phosphate prodrug strategies aimed at improving the pharmacokinetic properties and enhancing the efficacy of GEM. In the study, a novel cyclic phosphate prodrug GEM derivative, 18c, was designed and synthesized. The prodrug exhibited significant antiproliferative activity in a variety of cancer cell lines, with an IC50 value of 3.6−19.2 nM, which was significantly higher than that of the positive control NUC-1031. The metabolic pathway of compound 18C reveals its bioactive metabolites to prolong anti-tumor effects. In addition, the two P chiral diastereomers isolated for the first time showed similar cytotoxicity and metabolic properties. In the 22Rv1 and BxPC-3 tumor models, 18C demonstrated significant in vivo anti-tumor effects, indicating its potential as a drug candidate for the treatment of castration-resistant prostate cancer and pancreatic cancer.
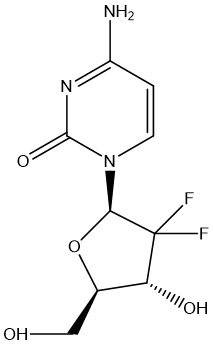
Figure 1. GEM
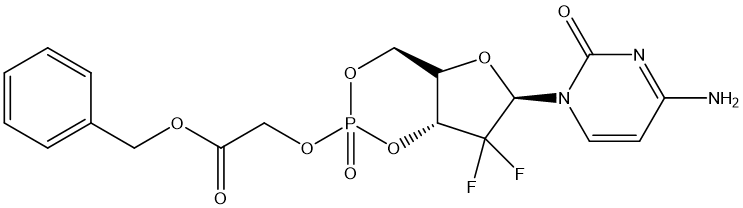
Figure 2. 18C
This study highlights the application potential of phosphate prodrug technology in improving drug efficacy and selectivity and sprovides a scientific basis for the development of new anticancer drugs.
1.2. Ketone prodrug
Ketone prodrug design aims to optimize the pharmacokinetic and pharmacodynamic properties of an active drug molecule by converting it into a derivative containing a ketone group. The introduction of ketone groups not only enhances the bioavailability of the drug, facilitates the absorption of the drug by improving its solubility, but also enables the controlled release of the active drug through its reduction or other chemical transformation in a specific environment in vivo. This transformation is usually carried out under the catalysis of enzymes, or through other chemical reactions, which ensure the specific activation of the drug in specific tissues or cells, effectively improving targeting and reducing systemic side effects. Marina Pisano et al. synthesized a series of C2-symmetric hydroxylated biphenyl derivatives with the key structural feature of α, β-unsaturated ketones and were designed to develop drug candidates for malignant melanoma [16]. This ketone derivative can Michael's reaction with nucleophiles in cancer cells. Improving the delivery efficiency and biological activity of compounds provides valuable information for the development of new anticancer drugs. And plus, the slow conversion rate of ketone prodrugs helps to prolong the effective concentration maintenance time of the drug in the body, thereby reducing the frequency of administration, while maintaining the prodrug state in non-target areas, reducing the toxicity and side effects to normal tissues. Overall, the design strategy of ketone prodrugs provides an effective solution for improving treatment efficacy and patient compliance by finely regulating the solubility, stability, targeting, absorption, distribution, metabolism, and excretion processes of drugs.
One type of ketone prodrug example is Nabumetone. Nabumetone [17] is a nonsteroidal anti-inflammatory drug (NSAID) used to treat osteoarthritis and rheumatoid arthritis. Nabumetone is converted in vivo to 6MNA, a cyclooxygenase (COX) inhibitor with anti-inflammatory properties. The study found that nabumetone had anti-inflammatory and potentially joint-protective effects on the inflammatory response of SFs compared to 6MNA, while 6MNA, like other COX inhibitors, had a pro-inflammatory effect on SFs. 6MNA depletes prostaglandin E1 (PGE1) and stimulates the activation of Erk (extracellular signal-regulated kinase) upon prolonged exposure, while nabumetone inhibits Erk activation. 6MNA stimulates the secretion of MMP-1, while nabumetone inhibits the secretion of matrix metalloproteinase-1 (MMP-1), but not MMP-13. UO126 (Erk pathway inhibitor) and nabumetone inhibit NF-κB activation, while 6MNA enhances NF-κB activation. Nabumetone shows anti-inflammatory effects and may counteract the potential adverse effects of COX-2 inhibition.
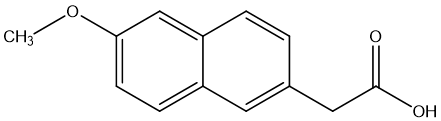
Figure 3. 6-methoxy-2-naphthylacetic acid(6MNA)
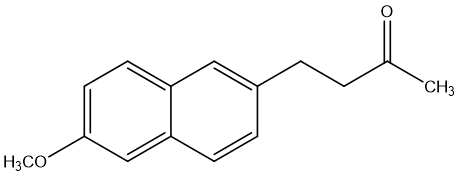
Figure 4. Nabumetone
The design and application of ketone prodrugs demonstrates how medicinal researchers can improve the therapeutic potential of drugs, reduce side effects, and optimize the therapeutic window of drugs through fine molecular design.
1.3. Ester prodrug
Esterification is a modification strategy widely used in drug discovery that involves reacting to polar groups in a drug molecule, such as carboxylic acids, hydroxyl groups, or amino groups, with alcohols, acids, or other suitable functional groups to form ester groups. This chemical modification significantly alters the physical, chemical, and biological properties of the drug. The main benefits of esterification include improved lipid solubility and membrane permeability of the drug [18], which makes it easier for the drug to penetrate cell membranes, especially when administered orally, improving drug absorption and distribution. In some cases, esterified prodrugs can effectively overcome tumor cell resistance to certain drugs, such as monochalcoplatin, which is a unilateral esterified tetravalent platinum prodrug containing chalcone that exhibits higher cytotoxicity and antitumor activity than cisplatin, while effectively overcoming cisplatin resistance [19]. Besides, esterification can prolong the duration of action of the drug, by slowing down the release rate of the drug in the body, so that the drug is gradually converted into the active form in the body. Esterification also helps to improve the targeting of the drug, allowing the drug to reach the target tissue or cell more precisely, with less impact on non-target areas [20]. For instance, the design of esterified prodrugs can also take advantage of the distribution of specific enzymes in specific tissues and substrate specificity, such as carboxylesterase (CES) in the liver and intestine, to promote the hydrolysis of the prodrug at specific sites, thereby improving drug efficacy and reducing side effects. Through precise design, it is possible to predict and optimize the intestinal absorption of prodrugs, as well as to evaluate their first-pass hydrolysis effects under different species and age conditions [21]. At the same time, esterification reduces the toxicity and side effects of the drug, because the prodrug form is relatively stable in the blood circulation and is not activated until it reaches a specific environment, such as a specific pH or where a specific enzyme is present. In addition, esterification improves the drug stability of the drug, which is beneficial for the storage and transportation of the drug. Finally, esterification allows for the regulation of drug release, and ester bonds can be selectively hydrolyzed in specific environments to achieve timed or localized release of the drug, thereby optimizing drug efficacy and safety.
The production of benoesters [22] is a classic ester prodrug example, which consists of aspirin (acetylsalicylic acid) and acetaminophen linked by esterification. This design not only retains the antipyretic and analgesic effects of both components, but also significantly reduces the risk of direct irritation and injury to the gastrointestinal tract by aspirin, as benolate is slowly hydrolyzed into the original drug in the body, reducing local acidic irritation.
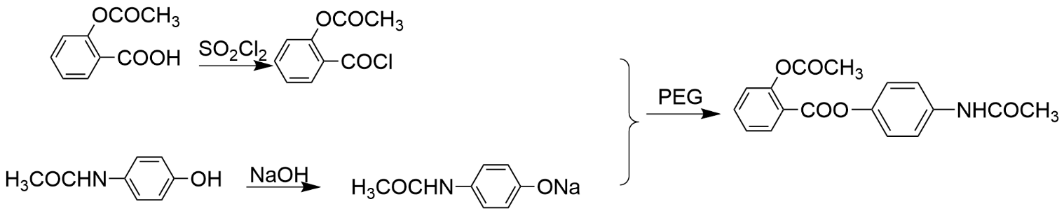
Figure 5. synthesis steps of benoxate
Through esterification modification, the lipophilicity of the drug is improved, the permeability of the drug is increased, and the oral absorption is improved. Pharmaceutical scientists are able to design safer, more effective treatments that meet clinical needs and optimize the patient’s experience.
1.4. Amidation prodrug
Amide bonds, covalent bonds formed by the reaction of carboxylic acids and amines, are often catalyzed in vivo by enzymes such as proteases, carboxylesterases, or peptidases, and the differences in the distribution of these enzymes in different tissues provide the possibility of targeted release of prodrugs. Although amide bonds are limited due to their high enzymatic stability in vivo and may affect the complete conversion of the prodrug to the active compound, in certain environments, such as cancer cells, amide prodrugs can be efficiently hydrolyzed through the action of specific enzymes [23], releasing the active drug, enabling targeted drug delivery and improved bioavailability. Furthermore, the incorporation of amide moieties substantially augments the molecular polarity, thereby facilitating aqueous solubility. This modification also exerts a salutary influence on the pharmacokinetic profile by enhancing ADME processes of the drug molecule. With prodrug design, the toxicity and side effects of the drug are reduced, as the prodrug itself is often low or inactive. In addition, by carefully designing the metabolic rate of the prodrug, the duration of action of the drug can be prolonged, the frequency of administration can be reduced, and the convenience of patients can be brought to patients. Overall, the design of amidated prodrugs provides a powerful chemical tool for improving drug efficacy, reducing side effects, and enabling precision medicine.
One typical example of the amidation prodrug is Levodopa (L-DOPA). Dopamine is an important neurotransmitter that plays a role in the brain and is associated with a variety of physiological functions such as mood, pleasure, reward mechanisms, and motor control. L-DOPA is a prodrug of dopamine that is commonly used to increase dopamine levels in the brain because dopamine itself cannot cross the blood-brain barrier. After L-DOPA enters the brain, it is taken up by dopaminergic neurons and converted into dopamine, thus exerting pharmacological effects. The amide L-DOPA (Carbidopa) [24] is a peripheral decarboxylase inhibitor, often used in conjunction with L-DOPA, with the aim of reducing the peripheral decarboxylation of L-DOPA into dopamine, thereby increasing the proportion of L-DOPA entering the brain through the blood-brain barrier, increasing dopamine concentration in the brain, and reducing peripheral side effects. As an amide prodrug, L-DOPA has significant advantages over dopamine in the treatment of Parkinson's disease, mainly in improving the penetration of the blood-brain barrier, enhancing in vivo stability, improving pharmacokinetic properties, reducing peripheral metabolism, achieving sustained drug release, and has the potential to reduce motor complications caused by long-term treatment. These properties make amide prodrugs show great potential to improve efficacy, reduce side effects, and optimize the patient experience.
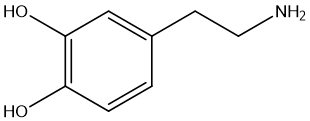
Figure 6 dopamine
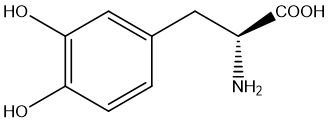
Figure 7 Levodopa
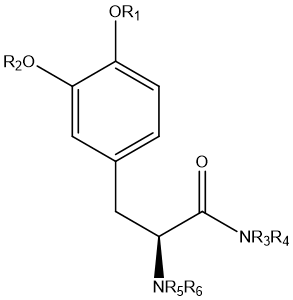
Figure 8 Chemical structures of general amide prodrugs
Amidation prodrugs are often designed to increase the polarity of the drug molecule, thereby increasing its solubility in water. Since amide bonds are relatively stable in living organisms, amidated prodrugs can be hydrolyzed by specific enzymes at the right time and place to release the active drug. This strategy is particularly suitable for those drug molecules that are inherently poorly water-soluble, improving the stability and solubility of the drug by forming amide bonds, and regulating the release rate of thedrug.
1.5. pH-responsive prodrug
pH-responsive prodrug is an intelligent drug delivery system that is designed with a specific chemical structure that triggers drug release in a specific pH environment. This property allows the drug to release the effective drug ingredient more precisely within the target site, such as tumor tissue or specific organelles (e.g., lysosomes). Tumor tissues typically have a low microenvironment pH [25], while certain organelles such as lysosomes also have an acidic environment, which provides ideal activation conditions for pH-responsive prodrugs. The advantage is that it not only enhances the targeting of the drug and reduces the impact on normal tissues, but also effectively reduces adverse reactions by reducing systemic drug exposure. In addition, pH-responsive prodrug enhance efficacy by accumulating the drug at the lesion site, increasing the local drug concentration. The intelligent release feature allows the drug to respond automatically according to physiological conditions, enabling efficient and adaptive treatment.
Dexamethasone prodrug [26] nanoparticles are an innovative pH-responsive drug delivery system that forms a novel prodrug by linking dexamethasone to a low molecular weight branched polyethylenimine. These prodrug nanoparticles exhibit sensitivity to a weakly acidic environment and are able to release drugs in response to acidic environments [27] such as inflammation, improving efficacy while reducing the impact on normal tissues. Its advantages include good biocompatibility, low cytotoxicity, stability, and efficient drug delivery, providing new strategies and possibilities for the treatment of respiratory diseases, including COVID-19.
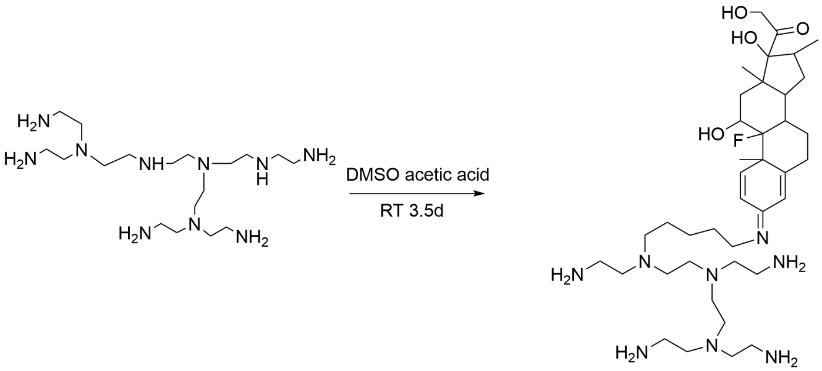
Figure 9. Flow chart of the synthesis of dexamethasone-polyethylenimine (DXM-PEIDP) prodrug nanoparticles
1.6. Enzyme activation prodrugs
The enzyme-activated prodrug strategy is an innovative approach to drug design that enhances the targeting of therapeutics and reduces side effects by designing drug molecules into prodrug forms to achieve conversion to the active form under the action of specific enzymes in the body. This strategy allows the drug to remain in a low or inactive state in non-target tissues, activated only by specific enzymes at the target location, effectively reducing damage to normal cells. Due to the lower systemic toxicity in the prodrug form, the risk of systemic side effects is reduced while improving efficacy. In addition, by achieving a high concentration of the drug at the site of the disease, the therapeutic effect can be enhanced, and the development of drug resistance may be slowed. Enzyme prodrug therapy (EPT) [28] further optimizes the pharmacokinetic properties of drugs through the development of well-designed enzyme prodrugs and self-immolative linkers (SIL), as well as long-cycle enzyme prodrugs, and provides an effective means to realize precision medicine.
Horseradish peroxidase (HRP) [29] and indole-3-acetic acid (IAA) systems: this is an example in a study that demonstrates the application of enzyme-activated prodrug strategies. In this system, horseradish peroxidase can be designed specifically recognized and catalyze the conversion of the prodrug indole-3-acetic acid (IAA) into an active drug molecule. IAA itself is non-toxic or low toxic, but once activated by HRP at the tumor site, it can be converted into a cytotoxic drug that kills tumor cells. HRP can be specifically delivered to the tumor site by antibody conjugation or gene delivery, ensuring therapeutic efficacy while limiting damage to healthy tissue.
Enzyme-activated prodrug technology uses the precise enzyme catalytic mechanism in organisms to realize the spatiotemporal controlled release of drugs and improve the safety and efficacy of treatment. This strategy is of particular interest in the field of anti-tumor therapeutics, as it enables more precise treatment by targeting the unique characteristics of the tumor microenvironment.
1.7. Carbamate prodrug
Carbamate prodrugs have effectively improved the pharmacokinetic properties of the drug by introducing the carbamate structure into the drug molecule, including enhancing the penetration of the biofilm, thereby increasing the bioavailability of the active drug in vivo [30]. This structural allows the drug to selectively break under specific conditions in vivo, such as pH changes or the presence of specific enzymes, to achieve controlled drug release, which is of great significance for achieving timed or localized drug delivery. In addition, the use of carbamate prodrugs also helps to reduce the distribution of the drug in non-target areas, reduce side effects, and enhance the storage and transportation capacity of the drug by improving the chemical stability of the drug. More advancedly, carbamate prodrugs can be directed to specific cells or tissues through targeted delivery systems, thereby improving efficacy while reducing systemic side effects, providing a more precise treatment option for drug therapy.
There are two examples about carbamate prodrugs. One is Prasugrel [31], an antiplatelet drug used to prevent thrombosis, is a new type of thienylpyridine prodrug, which belongs to the carbamate class prodrug. Prasugrel has shown a more significant reduction in cerebral infarction and peripheral arterial occlusive disease in multiple animal models, with effective doses ranging from 0.3 to 3 mg/kg. In clinical trials, prasugrel was shown to be more effective than clopidogrel in preventing ischemic events in patients with acute coronary syndromes. In addition, prasugrel has shown approximately 10 times more potency than clopidogrel in the treatment of peripheral arterial occlusive disease, suggesting that it may be more effective in the treatment of these conditions in the clinic.
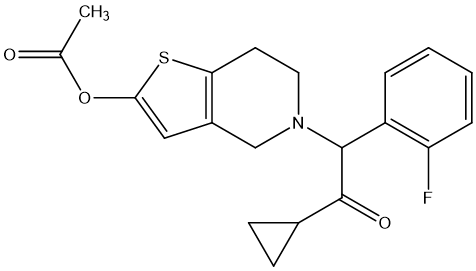
Figure 10. Prasugrel
The other is Curcumin [32], which is a polyphenolic compound with a variety of pharmacological effects, especially showing potential in alleviating diseases associated with neuroinflammation, including Alzheimer's disease, Parkinson's disease, multiple sclerosis, epilepsy, and brain damage. However, the therapeutic efficacy of curcumin is limited by its poor physicochemical properties, particularly its chemical and metabolic instability, resulting in extremely low oral bioavailability. As a novel prodrug of curcumin, curcumin diethyl γ-aminobutyrate (CUR-2GE) improves its stability in acidic environment and enhances lipophilicity through carbonate bond linkage, so that it can be rapidly converted into active curcumin under physiological pH conditions, showing higher cell uptake rate and more significant anti-inflammatory effects. These improved physicochemical properties and biological activity make CUR-2GE have better potential in anti-neuroinflammation, providing a promising new strategy for the treatment of related neurodegenerative diseases.
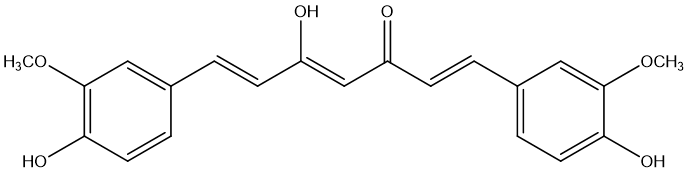
Figure 11. Curumin
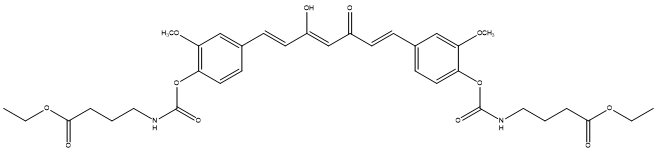
Figure 12. CUR-2GE
1.8. Liposomal Prodrugs
Liposomal prodrugs technology is a drug delivery system that combines the biocompatibility of liposomes with the translational properties of prodrugs. The technology uses tiny vesicles made up of phospholipid bilayers to mimic the structure of cell membranes to encapsulate water- or fat-soluble drugs. By coupling prodrug molecules inside or on the surface of liposomes, prodrug activation is realized in a specific physiological environment or under specific stimuli (such as enzymes, pH changes, light, etc.), and the parent drug with pharmacological activity is released. Liposomal prodrug technology has significant targeting, which can use the activation mechanism of tumor tissue-specific enzymes to promote the enrichment and activation of drugs at tumor sites, thereby reducing the toxic side effects on normal tissues [33]. In addition, the concentration of the active drug in the lesion after the transformation of the prodrug increased, which effectively improved the treatment efficiency. At the same time, the unactivated prodrug is relatively harmless, reducing systemic toxicity. The encapsulation of liposomes prolongs the half-life of the drug in the blood circulation, improves the stability of the drug, and improves its pharmacokinetic properties. Liposomal prodrug technology is also intelligently responsive [34], enabling the release of drugs according to specific physiological or pathological conditions for precise treatment. Besides, lipid-based formulations (LBFs), as a form of liposomal prodrug technology, are particularly suitable for oral administration and provide an effective solution to the oral absorption of hydrophobic drugs [35]. With the in-depth research on the working mechanism of LBF and the continuous advancement of related technologies, it is expected that LBF will play a more critical role in drug development and improvement. The development of solid-state LBF, which aims to improve stability and convenience, is one of the hot spots of current research. These advances show that liposomal prodrug technology has broad application prospects in the field of medicine and is expected to provide patients with safer and more effective treatment options.
One type of liposomal prodrug examples is Irinotecan [36], which is a potent topoisomerase I inhibitor that acts by conversion to the active metabolite SN-38, a process that involves the catalysis of carboxylesterase (CES) and ultimately inactivation by the action of cytochrome P450 3A4/3A5 (CYP3A4/5) and uridine diphosphate glucuronosyltransferase 1A (UGT1A) family enzymes. Differences in the expression and activity of these enzymes between individuals may affect the efficacy and toxicity of irinotecan. Liposomal irinotecan (nal-IRI) achieves longer-lasting SN-38 release through enhanced permeability and retention accumulation in tumor tissues compared to conventional non-liposomal irinotecan, thereby enhancing cytotoxicity. In addition, liposomal irinotecan benefits from the protective effect of liposomes, with a longer half-life and a higher area under the drug curve (AUC) in plasma, which helps to improve efficacy and reduce toxic side effects. Although liposomal irinotecan provides longer drug exposure, its overall toxicity is comparable to that of non-liposomal irinotecan, showing a better safety profile in clinical applications.
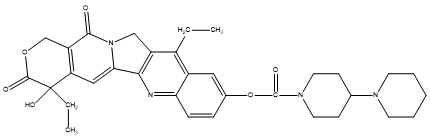
Figure 13. Irinotecan
1.9. Pleiotropic prodrugs
Pleiotropic prodrug aims to target multiple different biological targets or pathways simultaneously or sequentially through a single molecule, in order to enhance efficacy, reduce dose-dependent side effects, overcome drug resistance, and other issues. Such prodrugs usually contain two or more pharmacologically active units, which can be released in the body at the right time through enzymatic or non-enzymatic reactions to exert their respective or synergistic therapeutic effects. Pleiotropic prodrugs have great potential for the treatment of Alzheimer's disease, which can protect nerve activity, reduce side effects, and easily penetrate the blood-brain barrier. In addition, the multi-modal attack strategy of pleiotropic prodrug can help overcome the development of drug resistance, providing patients with a safer and more effective treatment plan, which can not only comprehensively intervene in the complex pathological process of the disease and enhance the treatment effect, but also effectively reduce adverse reactions and improve patients' treatment compliance by optimizing drug dosage and reducing non-target effects.
For instance, carbamate 7 [37] is an innovative pleiotropic prodrug designed to treat Alzheimer's disease by covalently inhibiting butyrylcholinesterase (BuChE) and subsequently releasing Carbamate 6, which has a 5-HT6 receptor antagonistic effect. This dual mechanism may improve efficacy, reduce side effects, and enhance drug targeting in the brain. Both in vitro and in vivo experimental results have shown that compound 7 has the potential to improve memory and cognitive function, making it a promising candidate in Alzheimer's disease treatment research.
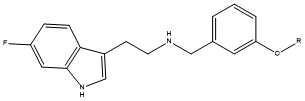
Figure 14. Carbamate 7 R=CON(ME)Et
The design and application of pleiotropic prodrugs reflect the in-depth understanding of disease complexity and the innovation of treatment strategies in drug development, and although there are challenges such as difficult design and complex safety assessment, they have great potential to improve treatment efficacy and improve patients' quality of life.
2. Latest developments and research in the field of prodrugs
Prodrug technology, as an important drug delivery strategy, has made significant progress in the past few years. These advances are reflected not only in the innovation of drug design and synthesis technology, but also in the use of prodrugs in clinical trials and the improvement of their treatment outcomes for patients.
Due to the special internal environment inside the tumor cells, there is no doubt that prodrugs have a very high level of popularity in the field of cancer treatment. The method of activating prodrugs through radiopharmaceuticals has become an effective way to treat cancer. For example, with the radiopharmaceutical [18F] FDG, the axial ligand release of the Pt(IV) complex can be triggered inside the tumor, a process that is achieved by a hydrated electron-mediated mechanism of water radiation cleavage [38]. This approach not only enhances the targeting of the drug, but also reduces systemic side effects. In addition, the cell viability curves observed in vitro experiments showed that when Pt-ADC interacted with [18F] FDG, the cell viability rate was close to that of MMAE treatment alone, indicating that Pt-ADC has good stability and efficient reactivity to [18F] FDG in living cells.
In terms of tumor-specific drug delivery, important progress has also been made in the design strategies of acid-sensitive prodrugs and specific fluorescent prodrugs. Acid-sensitive prodrugs take advantage of low pH conditions in the tumor microenvironment to control drug release through specific chemical bonds [39]. For example, some prodrugs that are designed to release drugs at pH 5 and stabilize at pH 7.4 have been shown to provide sustained drug release for more than 15 days and alleviate symptoms of inflammation in animal models. Specific fluorescent prodrugs, on the other hand, not only improve intracellular accumulation and bioavailability, but also report their localization and activation through real-time fluorescence changes [40]. These properties enable the efficient accumulation of prodrugs in tumor cells of different organs, providing a new perspective for tumor treatment. At the same time, glutathione-triggered prodrugs have also attracted attention due to their rapid release of active drugs in tumor cells due to the high glutathione concentration environment in tumor cells [41]. For example, polysulfide-based prodrug nanodrugs use high concentrations of glutathione in tumor cells for selective release, showing superior therapeutic potential [42]. These prodrugs enable the release of drugs within tumor cells through thiol-polysulfide exchange reactions, resulting in highly effective local therapeutic effects. The cancer cell delivery platform provides a new direction for precision drug delivery.
In addition to research in the field of cancer, prodrugs are also emerging in the field of nano and antimicrobial drugs. Modular lipid nanoparticle (LNPs) technology offers the possibility of efficient delivery of siRNAs and other nucleic acid drugs. The interchangeability of LNPs and their high compatibility with RNA payloads make them extremely flexible systems [43]. In addition, lipophilic small molecule prodrugs synthesized by drug modification methods can be stably bound to LNPs, providing an opportunity to co-encapsulate multiple therapeutics in the same formulation, thereby achieving the effect of combination therapy. For long-acting drug delivery systems, prodrug strategies also show great potential. By designing long-acting prodrugs (LA-prodrugs), combined with existing or emerging drug delivery technologies, long-term drug release can be achieved in vivo. These prodrugs are designed to focus on controlling the release rate of the drug through cleavable linkers, thereby prolonging the duration of the drug's action [44]. In addition, antimicrobial prodrugs to overcome bacterial resistance are also evolving [45]. Through drug reuse and derivatization, the application of nanotechnology, and the exploration of natural products, antimicrobial prodrugs can overcome the shortcomings of traditional drugs in terms of solubility, membrane permeability, chemical stability, etc., and improve the therapeutic effect.
Overall, the latest advances in prodrug technology provide new ideas for solving many challenges in traditional drug delivery. Whether it's to improve drug selectivity, enhance drug stability, or prolong drug action, prodrugs have demonstrated their unique advantages. Cheminformatics and data science analysis of approved prodrugs reveals trends in the development of prodrugs. The analysis showed that more than half of the prodrugs with annotated design targets were designed to improve bioavailability, particularly by increasing permeability. In addition, prodrugs that are currently in clinical trials are increasingly being used to address more complex delivery issues, such as the proportion of tissue-targeted prodrugs increasing to 26.5%, which is 5% higher than the proportion of approved prodrugs alone [3]. This suggests that the design of prodrugs is shifting from simply improving bioavailability to solving more complex drug delivery challenges. With the continuous deepening of research and the advancement of technology, it is expected that prodrugs will play a more important role in clinical practice in the future.
3. Conclusion
The above content describes a variety of chemical modification methods for prodrugs and discusses the application and clinical research of prodrugs in recent years. Each method has its own unique advantages, such as improved water solubility, increased bioavailability, enhanced stability, reduced irritation, modulated drug release, improved targeting, etc. Through the application of these approaches, drug scientists can design safer and more effective treatments that meet clinical needs and optimize the patient’s experience. For example, phosphate modifications can significantly increase the solubility of drug molecules in water, improve bioavailability, enhance stability, reduce irritation, and regulate drug release. Esterification can improve lipid solubility and membrane permeability, prolong the duration of action, and improve targeting. Amidation prodrugs increase water solubility and improve pharmacokinetic properties by introducing amide groups. pH-responsive prodrugs can trigger drug release at a specific pH environment, improving targeting and reducing side effects. Enzyme-activated prodrugs are converted into active drug molecules by the action of specific enzymes, improving targeting and reducing toxicity. Carbamate prodrugs improve ADME properties by releasing active drugs under specific conditions in vivo. Liposomal prodrugs combine liposomal technology and prodrug design to improve targeting and reduce toxicity. Pleiotropic prodrugs target multiple biological targets or pathways with a single molecule, enhancing therapeutic efficacy and reducing side effects.
In summary, these modification methods aim to optimize the pharmacokinetic properties and pharmacodynamic properties of drug molecules by changing their chemical structure, without changing their final therapeutic effect, and improve bioavailability and biofilm permeability, enhance targeting, prolong the drug action time, improve the water solubility and stability of drugs, reduce toxic side effects and adverse reactions, overcome undesirable odors or physicochemical properties, and meet the needs of the formulation. Therefore, prodrug design is a key step in optimizing the drug development process, which can not only improve the efficacy and safety of drugs, but also improve the treatment experience and compliance of patients, which is of great value to the field of drug development. At present, the future prospect of prodrug development is to develop in the direction of more precision, intelligence, personalization and environmental friendliness, through the application of cutting-edge technologies such as nanotechnology, computing-aided design, multidisciplinary integration and personalized medicine, to achieve efficient targeted delivery of drug molecules, intelligent response release and safety improvement, while focusing on green chemistry and ethical regulations, in order to ensure patient safety and improve efficacy, while promoting the sustainable development of the pharmaceutical industry.
References
[1]. Rautio, J., Kumpulainen, H., Heimbach, T., Oliyai, R., Oh, D., Järvinen, T., & Savolainen, J. (2008). Prodrugs: Design and clinical applications. Nature Reviews Drug Discovery, 7(4), 255–270. https://doi.org/10.1038/nrd2468
[2]. Huttunen, K. M., Raunio, H., & Rautio, J. (2011). Prodrugs: Design and clinical applications. Pharmacological Reviews, 63(3), 750–771. https://doi.org/10.1124/pr.110.003459
[3]. Fralish, Z., Chen, A., Khan, S., Zhou, P., & Reker, D. (2024). Machine learning in drug discovery. Nature Reviews Drug Discovery, 23(4), 365–380. https://doi.org/10.1038/s41573-023-00273-9
[4]. Alanazi, A. S., Miccoli, A., & Mehellou, Y. (2021). Advances in prodrug strategies. Journal of Medicinal Chemistry, 64(24), 16703–16710. https://doi.org/10.1021/acs.jmedchem.1c01176
[5]. Chang, Y., Yao, S., Chen, Y., Huang, J., Wu, A., Zhang, M., Xu, F., Li, F., & Huang, Y. (2019). Nanoplatforms for cancer therapy. Nanoscale, 11(2), 611–621. https://doi.org/10.1039/C8NR08510K
[6]. Chen, W. H., Lei, Q., Yang, C. X., Jia, H. Z., Luo, G. F., Wang, X. Y., Liu, G., Cheng, S. X., & Zhang, X. Z. (2015). Functional mesoporous silica nanoparticles for imaging and drug delivery. Small, 11(42), 5230–5242. https://doi.org/10.1002/smll.201500540
[7]. Kavanaugh, W. M. (2020). Recent advances in biological therapy. Expert Opinion on Biological Therapy, 20(2), 163–171. https://doi.org/10.1080/14712598.2020.1698701
[8]. Kong, F., Liang, Z., Luan, D., Liu, X., Xu, K., & Tang, B. (2016). Fluorescence spectroscopy for biomolecule detection. Analytical Chemistry, 88(11), 6450–6456. https://doi.org/10.1021/acs.analchem.6b01002
[9]. Wang, P., Gong, Q., Hu, J., Li, X., & Zhang, X. (2021). Targeted cancer therapy with prodrugs. Journal of Medicinal Chemistry, 64(1), 298–325. https://doi.org/10.1021/acs.jmedchem.0c01234
[10]. Xie, A., Hanif, S., Ouyang, J., Tang, Z., Kong, N., Kim, N. Y., Qi, B., Patel, D., Shi, B., & Tao, W. (2020). Precision nanomedicine for cancer immunotherapy. EBioMedicine, 56, 102821. https://doi.org/10.1016/j.ebiom.2020.102821
[11]. Song, H., Li, W., Qi, R., Yan, L., Jing, X., Zheng, M., & Xiao, H. (2015). Multi-responsive nanocarriers for drug delivery. Chemical Communications, 51(68), 11493–11495. https://doi.org/10.1039/C5CC03817G
[12]. Kleeb, S., Jiang, X., Frei, P., Sigl, A., Bezençon, J., Bamberger, K., Schwardt, O., & Ernst, B. (2016). Targeted drug delivery with glycomimetics. Journal of Medicinal Chemistry, 59(7), 3163–3182. https://doi.org/10.1021/acs.jmedchem.6b00045
[13]. Mehellou, Y., Rattan, H. S., & Balzarini, J. (2018). Prodrugs in antiviral therapy. Journal of Medicinal Chemistry, 61(6), 2211–2226. https://doi.org/10.1021/acs.jmedchem.7b00754
[14]. Ji, X., Wang, J., Zhang, L., Zhao, L., Jiang, H., & Liu, H. (2013). Exploring novel therapeutic agents. Journal Name Missing, 48(X), 621–634.
[15]. Zhang, L., Qi, K., Xu, J., Xing, Y., Wang, X., Tong, L., He, Z., Xu, W., Li, X., & Jiang, Y. (2023). Advances in medicinal chemistry research. Journal of Medicinal Chemistry, 66(X), 4150–4166. https://doi.org/10.1021/acs.jmedchem.3c00045
[16]. Dettori, M. A., Pisano, M., Rozzo, C., Delogu, G., & Fabbri, D. (2021). New chemical entities in cancer therapy. ChemMedChem, 16(7), 1022–1033. https://doi.org/10.1002/cmdc.202100056
[17]. Pillinger, M. H., Dinsell, V., Apsel, B., Tolani, S. N., Marjanovic, N., Chan, E. S., Gomez, P., Clancy, R., Chang, L. F., & Abramson, S. B. (2004). Inflammation pathways in rheumatoid arthritis. British Journal of Pharmacology, 142(5), 973–982. https://doi.org/10.1038/sj.bjp.0705840
[18]. Lavis, L. D. (2008). Fluorescent probes for biological imaging. ACS Chemical Biology, 3(4), 203–206. https://doi.org/10.1021/cb800025k
[19]. Ma, L., Wang, N., Ma, R., Li, C., Xu, Z., Tse, M. K., & Zhu, G. (2018). Catalytic chemistry for sustainable development. Angewandte Chemie International Edition, 57(X), 9098–9102. https://doi.org/10.1002/anie.201804113
[20]. Beaumont, K., Webster, R., Gardner, I., & Dack, K. (2003). Drug metabolism and disposition in pharmacokinetics. Current Drug Metabolism, 4(5), 461–485. https://doi.org/10.2174/1389200033489317
[21]. Ohura, K. (2020). Yakugaku Zasshi: Journal of the Pharmaceutical Society of Japan, 140(4), 369–376.
[22]. Wang, W., Lü, W., & Lu, Z. (2006). [Details needed for the journal title]. 39(42).
[23]. Park, Y., Park, J. H., Park, S., Lee, S. Y., Cho, K. H., Kim, D. D., Shim, W. S., Yoon, I. S., Cho, H. J., & Maeng, H. J. (2016). Molecules, 21(1).
[24]. Haddad, F., Sawalha, M., Khawaja, Y., Najjar, A., & Karaman, R. (2017). Molecules, 23(1).
[25]. Delatouche, R., Denis, I., Grinda, M., El Bahhaj, F., Baucher, E., Collette, F., Héroguez, V., Grégoire, M., Blanquart, C., & Bertrand, P. (2013). European Journal of Pharmaceutics and Biopharmaceutics, 85(4), 862–872.
[26]. Su, M., & Yin, Z. (2022). [Details needed for the journal title]. 37(1), 130–133.
[27]. Su, M., Yang, B., Xi, M., Qiang, C., & Yin, Z. (2021). Journal of Drug Delivery Science and Technology, 66, 102738.
[28]. Walther, R., Rautio, J., & Zelikin, A. N. (2017). Advanced Drug Delivery Reviews, 118, 65–77.
[29]. Liang, Q., Xi, J., Gao, X. J., Zhang, R., Yang, Y., Gao, X., Yan, X., Gao, L., & Fan, K. (2020). [Details needed for the journal title]. 35(1).
[30]. Li, Y., Wang, Y., Zhang, R., Liu, C., Wei, Y., Sun, J., He, Z., Xu, Y., & Zhang, T. (2018). Drug Delivery and Translational Research, 8(6), 1335–1344.
[31]. Ogawa, T., Hashimoto, M., Niitsu, Y., Jakubowski, J. A., Tani, Y., Otsuguro, K., Asai, F., & Sugidachi, A. (2009). European Journal of Pharmacology, 612(1–3), 29–34.
[32]. Jithavech, P., Suwattananuruk, P., Hasriadi, Muangnoi, C., Thitikornpong, W., Towiwat, P., Vajragupta, O., & Rojsitthisak, P. (2022). PLOS ONE, 17(3), e0265689.
[33]. Mura, S., Bui, D. T., Couvreur, P., & Nicolas, J. (2015). Journal of Controlled Release, 208, 25–41.
[34]. Markovic, M., Ben-Shabat, S., Keinan, S., Aponick, A., Zimmermann, E. M., & Dahan, A. (2018). Pharmaceutics, 10(4).
[35]. Feeney, O. M., Crum, M. F., McEvoy, C. L., Trevaskis, N. L., Williams, H. D., Pouton, C. W., Charman, W. N., Bergström, C. A. S., & Porter, C. J. H. (2016). Advanced Drug Delivery Reviews, 101, 167–194.
[36]. Milano, G., Innocenti, F., & Minami, H. (2022). Cancer Science, 113(8), 2224–2231.
[37]. Toublet, F. X., Lalut, J., Hatat, B., Lecoutey, C., Davis, A., Since, M., Corvaisier, S., Freret, T., Sopková-de Oliveira Santos, J., Claeysen, S., Boulouard, M., Dallemagne, P., & Rochais, C. (2021). European Journal of Medicinal Chemistry, 210, 113059.
[38]. Wang, C., Xu, M., Zhang, Z., Zeng, S., Shen, S., Ding, Z., Chen, J., Cui, X. Y., & Liu, Z. (2024). Science Bulletin.
[39]. Nazli, A., Irshad Khan, M. Z., Rácz, Á., & Béni, S. (2024). European Journal of Medicinal Chemistry, 276, 116699.
[40]. Ma, S., Kim, J. H., Chen, W., Li, L., Lee, J., Xue, J., Liu, Y., Chen, G., Tang, B., Tao, W., & Kim, J. S. (2023). Advanced Science.
[41]. Zhao, J., Li, X., Ma, T., Chang, B., Zhang, B., & Fang, J. (2024). Medicinal Research Reviews, 44(4), 1013–1054.
[42]. Wang, C., Sui, W., Chen, W., Zhang, Y., Xing, J., Jiang, H., Xu, W., & Xing, D. (2024). Coordination Chemistry Reviews, 519, 216138.
[43]. van der Meel, R., Chen, S., Zaifman, J., Kulkarni, J. A., Zhang, X. R. S., Tam, Y. K., Bally, M. B., Schiffelers, R. M., Ciufolini, M. A., Cullis, P. R., & Tam, Y. Y. C. (2021). Small, 17(51), e2103025.
[44]. Chien, S. T., Suydam, I. T., & Woodrow, K. A. (2023). Advanced Drug Delivery Reviews, 198, 114860.
[45]. Maria, C., de Matos, A. M., & Rauter, A. P. (2024). Pharmaceuticals, 17(1).
Cite this article
Li,J. (2024). Advances and applications of prodrug strategies in drug design. Journal of Food Science, Nutrition and Health,3,12-22.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Journal:Journal of Food Science, Nutrition and Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Rautio, J., Kumpulainen, H., Heimbach, T., Oliyai, R., Oh, D., Järvinen, T., & Savolainen, J. (2008). Prodrugs: Design and clinical applications. Nature Reviews Drug Discovery, 7(4), 255–270. https://doi.org/10.1038/nrd2468
[2]. Huttunen, K. M., Raunio, H., & Rautio, J. (2011). Prodrugs: Design and clinical applications. Pharmacological Reviews, 63(3), 750–771. https://doi.org/10.1124/pr.110.003459
[3]. Fralish, Z., Chen, A., Khan, S., Zhou, P., & Reker, D. (2024). Machine learning in drug discovery. Nature Reviews Drug Discovery, 23(4), 365–380. https://doi.org/10.1038/s41573-023-00273-9
[4]. Alanazi, A. S., Miccoli, A., & Mehellou, Y. (2021). Advances in prodrug strategies. Journal of Medicinal Chemistry, 64(24), 16703–16710. https://doi.org/10.1021/acs.jmedchem.1c01176
[5]. Chang, Y., Yao, S., Chen, Y., Huang, J., Wu, A., Zhang, M., Xu, F., Li, F., & Huang, Y. (2019). Nanoplatforms for cancer therapy. Nanoscale, 11(2), 611–621. https://doi.org/10.1039/C8NR08510K
[6]. Chen, W. H., Lei, Q., Yang, C. X., Jia, H. Z., Luo, G. F., Wang, X. Y., Liu, G., Cheng, S. X., & Zhang, X. Z. (2015). Functional mesoporous silica nanoparticles for imaging and drug delivery. Small, 11(42), 5230–5242. https://doi.org/10.1002/smll.201500540
[7]. Kavanaugh, W. M. (2020). Recent advances in biological therapy. Expert Opinion on Biological Therapy, 20(2), 163–171. https://doi.org/10.1080/14712598.2020.1698701
[8]. Kong, F., Liang, Z., Luan, D., Liu, X., Xu, K., & Tang, B. (2016). Fluorescence spectroscopy for biomolecule detection. Analytical Chemistry, 88(11), 6450–6456. https://doi.org/10.1021/acs.analchem.6b01002
[9]. Wang, P., Gong, Q., Hu, J., Li, X., & Zhang, X. (2021). Targeted cancer therapy with prodrugs. Journal of Medicinal Chemistry, 64(1), 298–325. https://doi.org/10.1021/acs.jmedchem.0c01234
[10]. Xie, A., Hanif, S., Ouyang, J., Tang, Z., Kong, N., Kim, N. Y., Qi, B., Patel, D., Shi, B., & Tao, W. (2020). Precision nanomedicine for cancer immunotherapy. EBioMedicine, 56, 102821. https://doi.org/10.1016/j.ebiom.2020.102821
[11]. Song, H., Li, W., Qi, R., Yan, L., Jing, X., Zheng, M., & Xiao, H. (2015). Multi-responsive nanocarriers for drug delivery. Chemical Communications, 51(68), 11493–11495. https://doi.org/10.1039/C5CC03817G
[12]. Kleeb, S., Jiang, X., Frei, P., Sigl, A., Bezençon, J., Bamberger, K., Schwardt, O., & Ernst, B. (2016). Targeted drug delivery with glycomimetics. Journal of Medicinal Chemistry, 59(7), 3163–3182. https://doi.org/10.1021/acs.jmedchem.6b00045
[13]. Mehellou, Y., Rattan, H. S., & Balzarini, J. (2018). Prodrugs in antiviral therapy. Journal of Medicinal Chemistry, 61(6), 2211–2226. https://doi.org/10.1021/acs.jmedchem.7b00754
[14]. Ji, X., Wang, J., Zhang, L., Zhao, L., Jiang, H., & Liu, H. (2013). Exploring novel therapeutic agents. Journal Name Missing, 48(X), 621–634.
[15]. Zhang, L., Qi, K., Xu, J., Xing, Y., Wang, X., Tong, L., He, Z., Xu, W., Li, X., & Jiang, Y. (2023). Advances in medicinal chemistry research. Journal of Medicinal Chemistry, 66(X), 4150–4166. https://doi.org/10.1021/acs.jmedchem.3c00045
[16]. Dettori, M. A., Pisano, M., Rozzo, C., Delogu, G., & Fabbri, D. (2021). New chemical entities in cancer therapy. ChemMedChem, 16(7), 1022–1033. https://doi.org/10.1002/cmdc.202100056
[17]. Pillinger, M. H., Dinsell, V., Apsel, B., Tolani, S. N., Marjanovic, N., Chan, E. S., Gomez, P., Clancy, R., Chang, L. F., & Abramson, S. B. (2004). Inflammation pathways in rheumatoid arthritis. British Journal of Pharmacology, 142(5), 973–982. https://doi.org/10.1038/sj.bjp.0705840
[18]. Lavis, L. D. (2008). Fluorescent probes for biological imaging. ACS Chemical Biology, 3(4), 203–206. https://doi.org/10.1021/cb800025k
[19]. Ma, L., Wang, N., Ma, R., Li, C., Xu, Z., Tse, M. K., & Zhu, G. (2018). Catalytic chemistry for sustainable development. Angewandte Chemie International Edition, 57(X), 9098–9102. https://doi.org/10.1002/anie.201804113
[20]. Beaumont, K., Webster, R., Gardner, I., & Dack, K. (2003). Drug metabolism and disposition in pharmacokinetics. Current Drug Metabolism, 4(5), 461–485. https://doi.org/10.2174/1389200033489317
[21]. Ohura, K. (2020). Yakugaku Zasshi: Journal of the Pharmaceutical Society of Japan, 140(4), 369–376.
[22]. Wang, W., Lü, W., & Lu, Z. (2006). [Details needed for the journal title]. 39(42).
[23]. Park, Y., Park, J. H., Park, S., Lee, S. Y., Cho, K. H., Kim, D. D., Shim, W. S., Yoon, I. S., Cho, H. J., & Maeng, H. J. (2016). Molecules, 21(1).
[24]. Haddad, F., Sawalha, M., Khawaja, Y., Najjar, A., & Karaman, R. (2017). Molecules, 23(1).
[25]. Delatouche, R., Denis, I., Grinda, M., El Bahhaj, F., Baucher, E., Collette, F., Héroguez, V., Grégoire, M., Blanquart, C., & Bertrand, P. (2013). European Journal of Pharmaceutics and Biopharmaceutics, 85(4), 862–872.
[26]. Su, M., & Yin, Z. (2022). [Details needed for the journal title]. 37(1), 130–133.
[27]. Su, M., Yang, B., Xi, M., Qiang, C., & Yin, Z. (2021). Journal of Drug Delivery Science and Technology, 66, 102738.
[28]. Walther, R., Rautio, J., & Zelikin, A. N. (2017). Advanced Drug Delivery Reviews, 118, 65–77.
[29]. Liang, Q., Xi, J., Gao, X. J., Zhang, R., Yang, Y., Gao, X., Yan, X., Gao, L., & Fan, K. (2020). [Details needed for the journal title]. 35(1).
[30]. Li, Y., Wang, Y., Zhang, R., Liu, C., Wei, Y., Sun, J., He, Z., Xu, Y., & Zhang, T. (2018). Drug Delivery and Translational Research, 8(6), 1335–1344.
[31]. Ogawa, T., Hashimoto, M., Niitsu, Y., Jakubowski, J. A., Tani, Y., Otsuguro, K., Asai, F., & Sugidachi, A. (2009). European Journal of Pharmacology, 612(1–3), 29–34.
[32]. Jithavech, P., Suwattananuruk, P., Hasriadi, Muangnoi, C., Thitikornpong, W., Towiwat, P., Vajragupta, O., & Rojsitthisak, P. (2022). PLOS ONE, 17(3), e0265689.
[33]. Mura, S., Bui, D. T., Couvreur, P., & Nicolas, J. (2015). Journal of Controlled Release, 208, 25–41.
[34]. Markovic, M., Ben-Shabat, S., Keinan, S., Aponick, A., Zimmermann, E. M., & Dahan, A. (2018). Pharmaceutics, 10(4).
[35]. Feeney, O. M., Crum, M. F., McEvoy, C. L., Trevaskis, N. L., Williams, H. D., Pouton, C. W., Charman, W. N., Bergström, C. A. S., & Porter, C. J. H. (2016). Advanced Drug Delivery Reviews, 101, 167–194.
[36]. Milano, G., Innocenti, F., & Minami, H. (2022). Cancer Science, 113(8), 2224–2231.
[37]. Toublet, F. X., Lalut, J., Hatat, B., Lecoutey, C., Davis, A., Since, M., Corvaisier, S., Freret, T., Sopková-de Oliveira Santos, J., Claeysen, S., Boulouard, M., Dallemagne, P., & Rochais, C. (2021). European Journal of Medicinal Chemistry, 210, 113059.
[38]. Wang, C., Xu, M., Zhang, Z., Zeng, S., Shen, S., Ding, Z., Chen, J., Cui, X. Y., & Liu, Z. (2024). Science Bulletin.
[39]. Nazli, A., Irshad Khan, M. Z., Rácz, Á., & Béni, S. (2024). European Journal of Medicinal Chemistry, 276, 116699.
[40]. Ma, S., Kim, J. H., Chen, W., Li, L., Lee, J., Xue, J., Liu, Y., Chen, G., Tang, B., Tao, W., & Kim, J. S. (2023). Advanced Science.
[41]. Zhao, J., Li, X., Ma, T., Chang, B., Zhang, B., & Fang, J. (2024). Medicinal Research Reviews, 44(4), 1013–1054.
[42]. Wang, C., Sui, W., Chen, W., Zhang, Y., Xing, J., Jiang, H., Xu, W., & Xing, D. (2024). Coordination Chemistry Reviews, 519, 216138.
[43]. van der Meel, R., Chen, S., Zaifman, J., Kulkarni, J. A., Zhang, X. R. S., Tam, Y. K., Bally, M. B., Schiffelers, R. M., Ciufolini, M. A., Cullis, P. R., & Tam, Y. Y. C. (2021). Small, 17(51), e2103025.
[44]. Chien, S. T., Suydam, I. T., & Woodrow, K. A. (2023). Advanced Drug Delivery Reviews, 198, 114860.
[45]. Maria, C., de Matos, A. M., & Rauter, A. P. (2024). Pharmaceuticals, 17(1).