







1. Introduction
Single-cell RNA sequencing (scRNA-seq) provides an exceptional opportunity to dissect cellular heterogeneity by capturing RNA information at the single-cell level and examining the differential expression of genes across individual cells . It surpasses bulk RNA sequencing, which averages gene expression over different types of cells in one tissue, potentially masking the gene expression dynamics occurring within specific cell subpopulations. In contrast, scRNA-seq excels in identifying variations in gene expression between individual cells, allowing for the precise determination of which genes are upregulated or downregulated across different cell types . Thus, scRNA-seq is particularly well-suited for the study of complex diseases, such as cancer, where cellular heterogeneity within the same tissue. Specifically, scRNA-seq can differentiate between various immune cell subtypes and reveal their functional roles in immune responses, shedding light on how the immune system interacts with tumor cells. Such insights are critical for understanding immune dysregulation in cancer and for advancing the development of targeted immunotherapies .
Bone marrow mononuclear cells (BMMCs), which comprise a population of single-nucleus cells extracted from the bone marrow, include hematopoietic stem cells, progenitor cells, and several mature cells such as lymphocytes and monocytes. These cells are closely associated with hematological disorders, including leukemia, lymphoma, and multiple myeloma, as well as certain immune-related diseases. Studying the changes in BMMCs under disease conditions can aid in identifying disease-specific biomarkers and potential therapeutic targets . As a result, BMMCs represent a critical focus of scRNA-seq research, particularly in the context of disease. However, within the scope of scRNA-seq analysis, cell annotation remains the most crucial and challenging step for accurately identifying cell functions. Determining cell types is often hindered by the limited availability of well-established biological markers and reference datasets, making it difficult to fully leverage the potential of scRNA-seq in functional characterization.
Current methods for cell annotation in scRNA-seq data can be broadly categorized into three approaches . The first category involves clustering-based annotation, where different clustering algorithms such as k-means or hierarchical clustering are applied to scRNA-seq data to group cells with similar expression patterns . However, while this approach can identify clusters of cells with analogous gene expression profiles, it does not directly reveal the specific cell types, and the simplicity of the method often results in lower accuracy. The second approach relies on known marker genes for specific cell types to annotate new single-cell datasets . While this method can be effective for well-characterized cell types, it is heavily dependent on the availability of established marker genes. As a result, it becomes challenging to annotate cell types or novel cell states for which no clear marker genes are known. The final approach applies supervised learning algorithms to predict cell types based on single-cell expression data. However, the application of scRNA-seq often requires careful consideration due to the high costs associated with single-cell sequencing, which limits the scope of data collection. Furthermore, scRNA-seq datasets vary significantly in sequencing depth, noise, and biological complexity, making it difficult to construct models that can be applied reliably across different datasets. Additionally, the inherent sparsity of scRNA-seq data, due to the low abundance of mRNA molecules and the failure to detect the expression of many genes, poses a risk of overfitting. Traditional machine learning methods, such as support vector machines (SVM) and random forests, have shown low performance in this situation. As the sequencing techniques developed, there is massive scRNA-seq data generated, which promises a good data basis for using deep learning to solve this challenge, making it highly suitable for scRNA-seq cell type classification.
In this paper, to address the aforementioned challenges, we utilized a dataset comprising raw RNA transcript read counts in bone marrow mononuclear cell subpopulations. We introduced ensemble learning into convolutional neural networks (CNNs), which are well-suited for handling high-dimensional data. Through convolutional operations, CNNs significantly reduce the dimensionality of data while minimizing the loss of valuable information. By incorporating ensemble learning, we were able to enhance the model's performance without drastically increasing the number of parameters, making it more efficient for analyzing sparse single-cell sequencing matrices. Additionally, we implemented regularization techniques and cross-validation during the training process to further optimize the model. To validate the effectiveness of our approach, we conducted comparative experiments with other classical deep learning models. In conclusion, our CNN-based ensemble model represents a step forward in addressing the challenges of cell type annotation in scRNA-seq data. Its strong performance across various metrics highlights the potential of deep learning to improve single-cell data analysis, paving the way for deeper insights into cellular heterogeneity and the mechanisms underlying complex diseases.
2. Materials and Methods
2.1. Datasets
We collected the public single cell data from an international competition, NeurIPS 2021: Multimodal Single-Cell Data Integration . All the raw data can be freely downloaded from the GEO database with accession number GSE194122. It contains single-cell multiomics data collected from bone marrow mononuclear cells of 12 healthy human donors. Half the samples were measured using the 10X Multiple Gene Expression and Chromatin Accessibility kit and half were measured using the 10X 3' Single-Cell Gene Expression kit with Feature Barcoding in combination with the BioLegend TotalSeq B Universal Human Panel v1.0. Samples were prepared using a standard protocol at four sites. The resulting data was then annotated to identify cell types and remove doublets. The dataset was designed with a nested batch layout such that some donor samples were measured at multiple sites with some donors measured at a single site. More detailed generation process information can be found at the GEO database. In total, it contains 10032 cells and 13822 genes and 14 different cell types. Using sklearn’s split function, split the single cell dataset and the labels into a training and test set. The training and test set contain 6688 and 3344 cells, respectively.
2.2. Methods
Model architecture: We developed a neural network architecture to predict single-cell bone marrow mononuclear cell types using scRNA-seq gene expression data. Our method comprises two key components. First, we individually implemented the multilayer perceptron (MLP), convolutional neural network (CNN), long short-term memory (LSTM), and attention-based models to establish baseline performance. The MLP serves as a fundamental feedforward neural network model, consisting of multiple fully connected layers, which is suitable for the simple data processing. The CNN, on the other hand, is particularly well-suited for handling high-dimensional gene expression data by exploiting local patterns within the data. Through convolutional operations, CNNs reduce the dimensionality of the input while retaining essential gene features, making them efficient for scRNA-seq analysis where the data is both sparse and high-dimensional. LSTM networks are a type of recurrent neural network (RNN) designed to capture long-range dependencies in sequential data. Their ability to retain information over extended time steps makes them highly effective in handling temporal dynamics, which can be beneficial when analyzing time-related gene expression patterns in scRNA-seq data. Finally, the Attention mechanism improves upon LSTMs by allowing the model to focus on the most relevant parts of the input sequence, which enables more effective learning of intricate relationships between genes. The second process is to use ensemble learning to combine the outputs of the MLP, CNN, and LSTM models, respectively. Ensemble learning is a strategy that integrates different algorithms into one metamodel to improve the prediction accuracy. In a voting ensemble learning, the outputs of several models, including MLP, CNN, and LSTM, were combined, respectively, to reduce the likelihood of poor generalization due to model bias, and lead to improved performance of single cell types identification . To further enhance the predictive performance of these models, we incorporated ensemble learning, a method designed to combine the strengths of multiple classifiers. Ensemble learning leverages the advantages of different models to produce a more accurate and robust final prediction of cell types in scRNA-seq data. By aggregating the outputs of different models, ensemble techniques can reduce the variance and mitigate the risk of overfitting, which is particularly valuable when working with sparse and noisy scRNA-seq data. In summary, by introducing ensemble learning, we achieved improved model accuracy and robustness, providing a powerful tool for the classification of bone marrow mononuclear cell types from scRNA-seq data.
2.3. Training approach
This section shows the experimental process and results of our model training. The model was implemented in the PyTorch 1.11 and Sklearn environment 1.0.2, using a combination of SGD and the Adam optimizer for efficient gradient descent optimization. Several hyperparameters were carefully tuned to optimize model performance. After multiple iterations of fine-tuning, the final configuration was determined as follows: the input feature size corresponds to the dimensionality of the scRNA-seq gene expression data, while the network architecture consists of 4 layers, allowing the model to capture complex interactions between genes. The output feature size is set to 14, corresponding to the 14 distinct cell types in the classification task. The minibatch size was set at 128 to ensure stable training without overwhelming memory resources, while still providing sufficient data for each training iteration. The initial learning rate was set to 0.0005, providing a balance between convergence speed and stability. To further refine training, a learning rate decay of 10% was applied every 200 iterations, which helped prevent overshooting the optimal solution and improved generalization. We believe this training process contributed to the model’s ability to efficiently classify bone marrow mononuclear cell types from scRNA-seq data.
2.4. Model Assessment
To assess our model for three-class classification, we employed important metrics: Accuracy (ACC) and Macro F1 Score, which used the terms true negative (TN), true positive (TP), false negative (FN), and false positive (FP).
\( Precision = \frac{TP}{TP+FP} \)
\( Recall = \frac{TP}{TP + FN} \)
\( F1 score = \frac{2 * Precision * Recall}{Precision + Recall} \)
\( Accuracy = \frac{TP + TN}{TP + FP + TN + FN} \)
3. Results
3.1. Compare different deep learning models for predicting Single-Cell Bone Marrow Mononuclear Cell Types
Initially, our model comprises three base architectures: MLP, CNN, and LSTM. The performance of the MLP model, due to its simplicity, struggled to capture key patterns and features from the single-cell data. Additionally, MLP is not well-suited for handling high-dimensional data, leading to suboptimal performance. The results for MLP were accuracy, F1 score, precision, and recall all around 0.7651, reflecting its limitations in extracting meaningful insights from the data. In contrast, the CNN and LSTM models showed improved performance. LSTM, which is specifically designed for sequential data such as DNA and protein sequences, did not perform as effectively on gene expression data, achieving an accuracy of approximately 0.8. While LSTM excels at capturing temporal dependencies, its application to gene expression data is less optimal. However, when combined with the attention mechanism, which enhances the model's ability to focus on important features, the performance of LSTM improved significantly, reaching an accuracy of around 0.89. Nevertheless, this was still slightly lower than the CNN model. Finally, the CNN model, due to its convolutional mechanism, is particularly well-suited for processing high-dimensional gene expression data. CNNs can efficiently reduce the dimensionality of tens of thousands of features through successive convolutional layers without losing critical information. The features extracted by the CNN are highly representative of cell types, resulting in strong classification performance. The CNN achieved an accuracy, F1 score, precision, and recall of 0.904, indicating its effectiveness in capturing the complex patterns within single-cell RNA-seq data.
Next, we introduced ensemble learning, a method that combines the strengths of multiple models to enhance overall performance. To maintain a lightweight architecture, we focused on integrating the LSTM and CNN models. The results show that, while the LSTM base model initially performed suboptimally, ensemble learning improved its performance significantly, raising accuracy to around 0.88, a notable 6-point increase. The CNN model, already performing well on its own, further benefited from ensemble learning, achieving an accuracy, F1 score, precision, and recall of 0.9143. This represents the best performance among all models tested, demonstrating the effectiveness of the ensemble approach in optimizing CNN's ability to handle high-dimensional single-cell RNA-seq data.
Table 1: Model performance
Algorithms | Precision | Recall | F1 | Accuracy |
MLP | 0.7651 | 0.7651 | 0.7651 | 0.7651 |
CNN | 0.904 | 0.904 | 0.904 | 0.904 |
CNN Ensemble | 0.9143 | 0.9143 | 0.9143 | 0.9143 |
LTSM | 0.8146 | 0.8146 | 0.8146 | 0.8146 |
LTSM ATT | 0.8983 | 0.8983 | 0.8983 | 0.8983 |
LTSM Ensemble | 0.8797 | 0.8797 | 0.8797 | 0.8797 |
CNN LTSM | 0.8704 | 0.8704 | 0.8704 | 0.8704 |
Further analysis was conducted to evaluate the prediction accuracy across all 14 distinct cell types. As shown in the accompanying figures and tables, the model demonstrates superior performance in classifying the various cell types. These results indicate that our approach consistently outperforms other methods, providing accurate and reliable predictions across different cell populations.
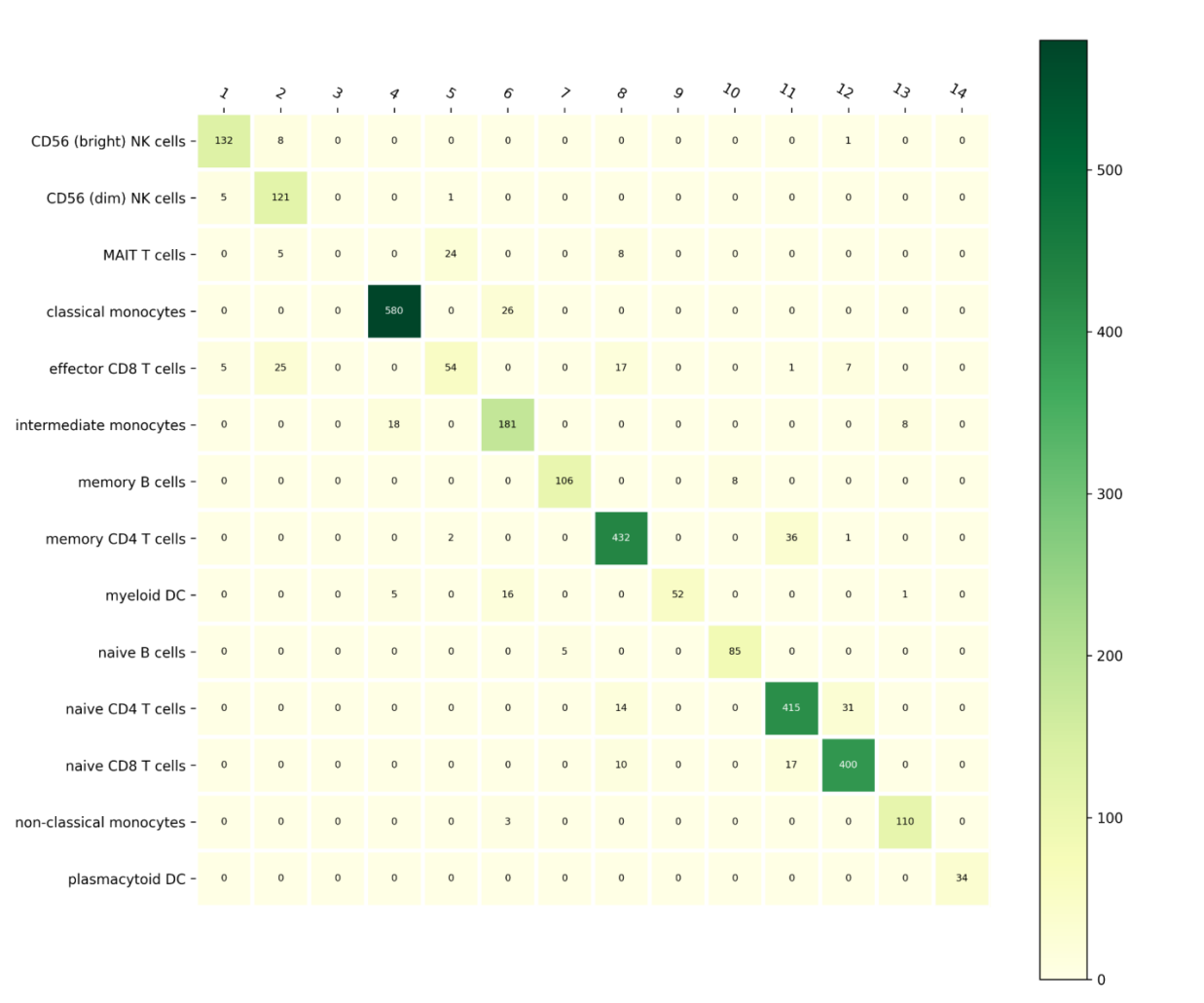
Figure 1: Cell Type Identification based on Single-cell RNA-seq
3.2. UMAP Visualization for different cell type annotations
Overall, the 14 cell types are clearly distinguishable, with particularly high accuracy in separating classical monocytes, memory CD4 T cells, and intermediate monocytes, as evidenced by the results. In contrast, naive CD8 T cells were more challenging to classify, with some overlap observed between this cell type and memory CD4 T cells. In summary, despite minor classification difficulties with certain cell types, the overall performance of the model was highly satisfactory, demonstrating strong predictive capability across the majority of cell types.

Figure 2: UMAP Visualization for different cell type annotations:
4. Conclusion
In this study, we introduced an ensemble learning-based CNN to achieve high-performance classification of single-cell bone marrow mononuclear cell types based on their gene expressions. Specifically, we began by collecting public single-cell data from the NeurIPS 2021: Multimodal Single-Cell Data Integration competition and preprocessed it for further analysis. Subsequently, we constructed several classical deep learning models, including MLP, LSTM, CNN, and attention-based architectures, all of which demonstrated strong performance, with the accuracy of more than 0.8. To further improve the model’s accuracy, we employed ensemble learning to combine homogeneous classifiers, like CNN, MLP and LSTM. The Comparative experiments revealed that the 1D CNN model with ensemble learning outperformed the other models, achieving an accuracy of 0.9143 and an F1 score of 0.9143. Finally, we utilized a heatmap to examine the classification results across different cell types, showing that our model provided highly accurate cell type annotations. Additionally, we also employed UMAP for visualization, which effectively illustrated the model’s classification performance.
In conclusion, our approach not only offers a powerful tool for the classification of single-cell bone marrow mononuclear cell types but also provides a robust solution for other single-cell annotation challenges. By leveraging a lightweight ensemble learning framework, we enhanced the effectiveness of cell type classification models, offering broader applications for single-cell data analysis.
Data and code availability
Datasets analyzed in this study are publicly available from: GEO (www.ncbi.nlm.nih.gov/geo/, with GSE194122). Trained model, additional documentation and code for training and predicting with our model available at: https://github.com/cclaireshi/Single_Cell_Classification_with_CNN-ensemble_Neural_Network
References
[1]. Brendel M, Su C, Bai Z, et al. Application of Deep Learning on Single-Cell RNA Sequencing Data Analysis: A Review. Genomics Proteomics Bioinformatics 2022; 20:814–835
[2]. Kuksin M, Morel D, Aglave M, et al. Applications of single-cell and bulk RNA sequencing in onco-immunology. Eur J Cancer 2021; 149:193–210
[3]. Ziegenhain C, Vieth B, Parekh S, et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol Cell 2017; 65:631-643.e4
[4]. Bao S, Li K, Yan C, et al. Deep learning-based advances and applications for single-cell RNA-sequencing data analysis. Brief Bioinform 2022; 23:
[5]. Wang T, Bai J, Nabavi S. Single-cell classification using graph convolutional networks. BMC Bioinformatics 2021; 22:364
[6]. Li S, Guo H, Zhang S, et al. Attention-based deep clustering method for scRNA-seq cell type identification. PLoS Comput Biol 2023; 19:e1011641
[7]. Wang X, Wang H, Liu D, et al. Deep learning using bulk RNA-seq data expands cell landscape identification in tumor microenvironment. Oncoimmunology 2022; 11:
[8]. Nguyen QT, Thanh LN, Hoang VT, et al. Bone Marrow-Derived Mononuclear Cells in the Treatment of Neurological Diseases: Knowns and Unknowns. Cell Mol Neurobiol 2023; 43:3211–3250
[9]. Huang H, Liu C, Wagle MM, et al. Evaluation of deep learning-based feature selection for single-cell RNA sequencing data analysis. Genome Biol 2023; 24:259
[10]. Flores M, Liu Z, Zhang T, et al. Deep learning tackles single-cell analysis—a survey of deep learning for scRNA-seq analysis. Brief Bioinform 2022; 23:
[11]. Lee J, Kim S, Hyun D, et al. Deep single-cell RNA-seq data clustering with graph prototypical contrastive learning. Bioinformatics 2023; 39:
[12]. Zhang X, Chen Z, Bhadani R, et al. NISC: Neural Network-Imputation for Single-Cell RNA Sequencing and Cell Type Clustering. Front Genet 2022; 13:
[13]. Jia S, Lysenko A, Boroevich KA, et al. scDeepInsight: a supervised cell-type identification method for scRNA-seq data with deep learning. Brief Bioinform 2023; 24:
[14]. Jiao L, Ren Y, Wang L, et al. MulCNN: An efficient and accurate deep learning method based on gene embedding for cell type identification in single-cell RNA-seq data. Front Genet 2023; 14:
[15]. Song T, Dai H, Wang S, et al. TransCluster: A Cell-Type Identification Method for single-cell RNA-Seq data using deep learning based on transformer. Front Genet 2022; 13:
[16]. Dong X, Chowdhury S, Victor U, et al. Semi-Supervised Deep Learning for Cell Type Identification From Single-Cell Transcriptomic Data. IEEE/ACM Trans Comput Biol Bioinform 2023; 20:1492–1505
[17]. Zhou Y, Peng M, Yang B, et al. scDLC: a deep learning framework to classify large sample single-cell RNA-seq data. BMC Genomics 2022; 23:504
[18]. Luecken MD, Burkhardt DB, Cannoodt R, et al. A sandbox for prediction and integration of DNA, RNA, and protein data in single cells. 2021;
[19]. Petegrosso R, Li Z, Kuang R. Machine learning and statistical methods for clustering single-cell RNA-sequencing data. Brief Bioinform 2020; 21:1209–1223
[20]. Xu F, Wang S, Dai X, et al. Ensemble learning models that predict surface protein abundance from single-cell multimodal omics data. Methods 2021; 189:65–73
Cite this article
Shi,C. (2024). A CNN-Ensemble-Based Neural Network for Enhanced Classification of Single-Cell Bone Marrow Mononuclear Cell Types. Applied and Computational Engineering,115,200-206.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 5th International Conference on Signal Processing and Machine Learning
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Brendel M, Su C, Bai Z, et al. Application of Deep Learning on Single-Cell RNA Sequencing Data Analysis: A Review. Genomics Proteomics Bioinformatics 2022; 20:814–835
[2]. Kuksin M, Morel D, Aglave M, et al. Applications of single-cell and bulk RNA sequencing in onco-immunology. Eur J Cancer 2021; 149:193–210
[3]. Ziegenhain C, Vieth B, Parekh S, et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol Cell 2017; 65:631-643.e4
[4]. Bao S, Li K, Yan C, et al. Deep learning-based advances and applications for single-cell RNA-sequencing data analysis. Brief Bioinform 2022; 23:
[5]. Wang T, Bai J, Nabavi S. Single-cell classification using graph convolutional networks. BMC Bioinformatics 2021; 22:364
[6]. Li S, Guo H, Zhang S, et al. Attention-based deep clustering method for scRNA-seq cell type identification. PLoS Comput Biol 2023; 19:e1011641
[7]. Wang X, Wang H, Liu D, et al. Deep learning using bulk RNA-seq data expands cell landscape identification in tumor microenvironment. Oncoimmunology 2022; 11:
[8]. Nguyen QT, Thanh LN, Hoang VT, et al. Bone Marrow-Derived Mononuclear Cells in the Treatment of Neurological Diseases: Knowns and Unknowns. Cell Mol Neurobiol 2023; 43:3211–3250
[9]. Huang H, Liu C, Wagle MM, et al. Evaluation of deep learning-based feature selection for single-cell RNA sequencing data analysis. Genome Biol 2023; 24:259
[10]. Flores M, Liu Z, Zhang T, et al. Deep learning tackles single-cell analysis—a survey of deep learning for scRNA-seq analysis. Brief Bioinform 2022; 23:
[11]. Lee J, Kim S, Hyun D, et al. Deep single-cell RNA-seq data clustering with graph prototypical contrastive learning. Bioinformatics 2023; 39:
[12]. Zhang X, Chen Z, Bhadani R, et al. NISC: Neural Network-Imputation for Single-Cell RNA Sequencing and Cell Type Clustering. Front Genet 2022; 13:
[13]. Jia S, Lysenko A, Boroevich KA, et al. scDeepInsight: a supervised cell-type identification method for scRNA-seq data with deep learning. Brief Bioinform 2023; 24:
[14]. Jiao L, Ren Y, Wang L, et al. MulCNN: An efficient and accurate deep learning method based on gene embedding for cell type identification in single-cell RNA-seq data. Front Genet 2023; 14:
[15]. Song T, Dai H, Wang S, et al. TransCluster: A Cell-Type Identification Method for single-cell RNA-Seq data using deep learning based on transformer. Front Genet 2022; 13:
[16]. Dong X, Chowdhury S, Victor U, et al. Semi-Supervised Deep Learning for Cell Type Identification From Single-Cell Transcriptomic Data. IEEE/ACM Trans Comput Biol Bioinform 2023; 20:1492–1505
[17]. Zhou Y, Peng M, Yang B, et al. scDLC: a deep learning framework to classify large sample single-cell RNA-seq data. BMC Genomics 2022; 23:504
[18]. Luecken MD, Burkhardt DB, Cannoodt R, et al. A sandbox for prediction and integration of DNA, RNA, and protein data in single cells. 2021;
[19]. Petegrosso R, Li Z, Kuang R. Machine learning and statistical methods for clustering single-cell RNA-sequencing data. Brief Bioinform 2020; 21:1209–1223
[20]. Xu F, Wang S, Dai X, et al. Ensemble learning models that predict surface protein abundance from single-cell multimodal omics data. Methods 2021; 189:65–73