







1. Introduction
1.1. Introduction of Astellolide S
Astellolides are a kind of sesquiterpenoids with several rare structure or functional groups, such as Astellolide R, which feature an special cage-like pentacyclic ring system. According to the research of Guangzhi Dai et al, chosen molecule Astellolide S has been confirmed to have the chemical formula C26H30O7 and was presumed to be a colorless needle-like crystal [1]. Importantly, Astellolide S possesses a very rare nicotinic acid building block, linked to C-6 through the ester linkage. It also possesses two ester groups, one γ-lactone and four chiral carbon atoms. So far, in figure 1, it seems that effective synthesis route has not been found for this molecule, but its special and reactive functional groups can be used as a breakthrough point for retrosynthesis analysis and synthesis analysis.
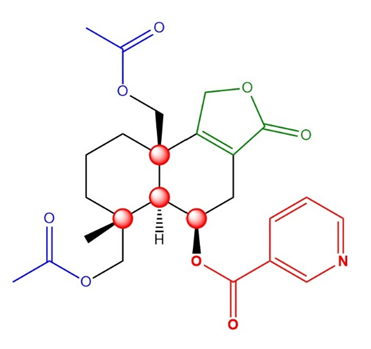
1.2. Source
Astellolide S is a natural product, which comes from an insect-derived fungi named Aspergillus parasiticus SDU001, associated with an arthropod, Armadillidium vulgare which is shown in figure 2 . Nowadays, this kind of fungi gets more and more attention because of its special role in producing bioactivity products. Guangzhi Dai et al.'s study used the OSMAC strategy to cultivate Aspergillus parasitic in Czapek-Dox medium, Martin medium, malt extract medium and rice medium, and used liquid chromatography-mass spectrometry analysis of ethyl acetate extracts to extract and identify new sesquiterpenoid Astellolide S from Aspergillus parasitic in rice medium [1].

1.3. Astellolide S and Astellolides
Up to now, 16 species of Astellolides has been reported in papers, namely Astellolide A-I, Astellolide Q-W, which are displayed in Figure 3. Among them, in the study of Tao Chen et al, it was proposed that Astellolide A-C and Astellolide Q extracted from marine fungi was bioactive, especially showing significant antibacterial activity [2]. Especially, Astellolide Q has a cage-like structure that is not common in natural products, while Astellolide S has a rare nicotinic acid as mentioned earlier.
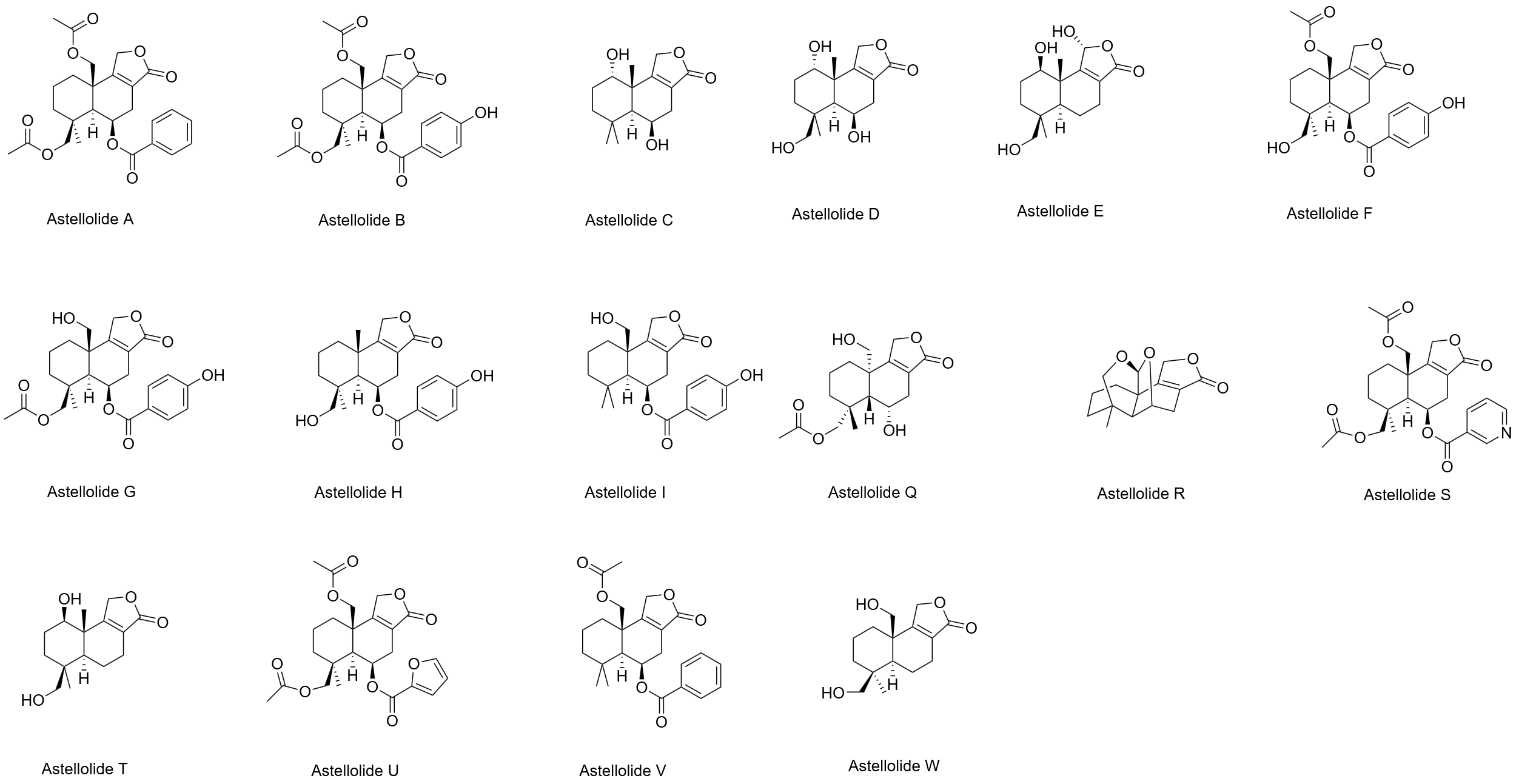
1.4. Biosynthesis and bioactivity
Yasutomo Shinohara et al. identified a novel sesquiterpene biosynthetic route and the gene clusters in 2016, successfully revealing the mechanism of Astellolide A and Astellolide B formation in Aspergillus oryzae [3]. Their findings are shown in figure 4.
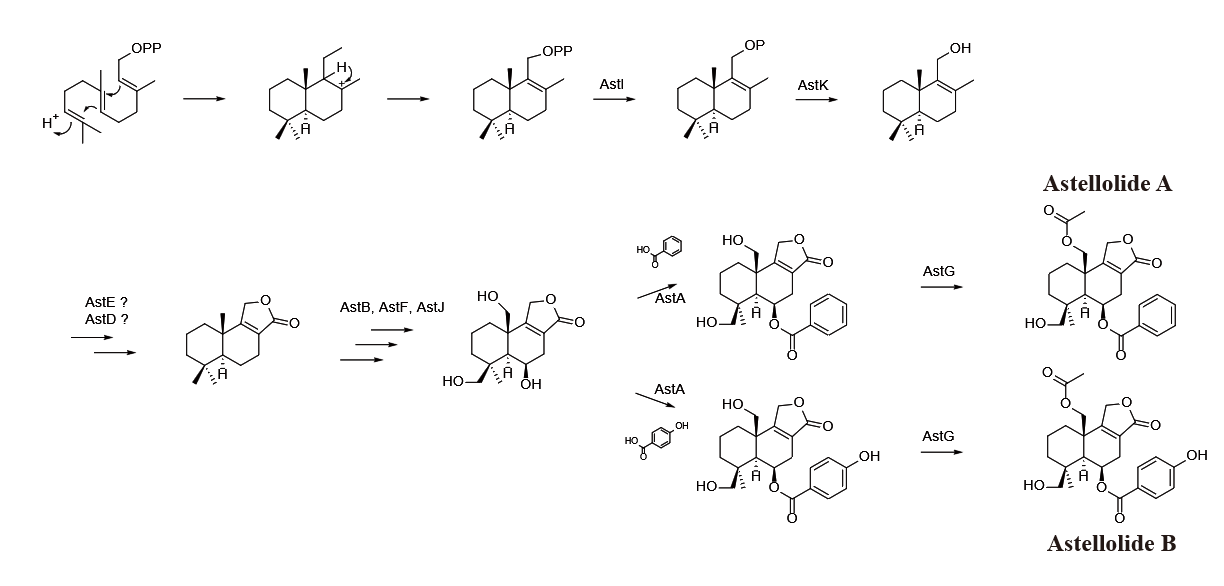
Based on enzyme reaction analysis, the research group discovered several enzymes (AstA, AstK, etc.). Although the molecule we chose is different from the ones studied in this study, they have the same carbon skeleton. The S-shaped initial molecule has the advantage of undergoing a cascade reaction to close the decalin system in less steps. Therefore, this biosynthetic route has an enlightment on how we could synthesize Astellolide S through absolute chemical methods.
2. Retrosynthesis and synthesis route 1
2.1. Retrosynthesis 1
Retrosynthesis route 1 simplifies molecules through retrosynthesis analysis by transforming a complex three-ring system into a core five-membered ring and side-chain compound through a series of reactions (including side chain dismantling, hydrolysis reaction, etc.) which is demonstrated in figure 5. In short, the core part of the route is to cut off carbon atoms 5 and 10 and carbon-carbon double bonds.
First, in figure 6, the reverse synthesis analysis of Astellolide S is carried out, starting from molecule 2, and the reverse deduction was carried out according to the reaction: the final product containing free hydroxyl group and TBSO protection group could be traced back to the precursor containing nicotinic acid building block and the corresponding position of hydroxyl group through the reverse process of TBSO protection group deprotection. The precursor continues to be pushed backwards, and the TBSO protective group is reversely eliminated, returning to the structure where the hydroxyl group is not protected by TBS, and the free hydroxyl group of this structure can be replenished by reverse hydrolysis of acyl group protection, and return to the compound containing acetoxy group and nicotinic acid building block. The compound is further reversed, and the multi-step acylation protection reaction is integrated, returning to molecule 1, using the "introduction-deprotection" reversibility of the protective group to precisely regulate the reactivity of functional groups such as hydroxyl groups.

Secondly, as it is shown in figure 7, the molecule 4 contains a red labeled structure, which can be reversed into the reverse process of esterification reaction between carboxyl groups and hydroxyl groups in intermediate products, and regress to intermediate structures containing free carboxyl groups and hydroxyl groups. Based on the formation reaction of the lactone ring (molecule 3), the lactone ring is eliminated in reverse, the ring-opening precursor containing carboxyl groups and hydroxyl groups is traced back to the ring-opening precursor containing carboxyl groups and hydroxyl groups, and the penta-membered cyclic ester is constructed through lactonization, and then the forward synthesis logic of the red label structure is introduced through esterification, and the reversibility of functional group reaction is used to clearly present the reverse synthesis and dismantling path of "lactone ring construction - esterification modification", which provides a reverse design basis for the functional group transformation and cyclization strategy in forward synthesis.

Then, molecule 8 in figure 8 can be traced back to the precursor molecule 7 containing ketone structure, double bonds and carboxylic acid side chains through the reverse disassembly ring reaction, and the precursor in which ketone can be reversely hydrolyzed, corresponding to the precursor of the double bond and carboxylic acid side chain of molecule 6. Further, the double-bond precursor can be reversed to the reverse process of hydroxyl addition and elimination, returning to the initial structure of the side chains containing hydroxyl groups, carbonyl groups and carboxylic acids, while the complex molecules containing polyfunctional groups in molecule 5 can be changed into the molecule containing the TBS protective group by double bond formation.

In addition, figure 9 illustrates that the simple molecules containing the chiral center, ethyl ester group, and carbonyl group are condensed by aldol reaction and the resulting hydroxyl group is protected to obtain a molecule containing a TBSO protective group. Then, a hydroxyl group is introduced at the α-carbon of the carbonyl group by reacting with formaldehyde [4]. Structures containing ethyl ester groups can form the original cyclic structure. And the carbonyl group of the side chain is protected to ensure the selectivity of the Wittig reaction described above. The overall backward logic revolves around "functional group reversal", gradually breaking down complex molecules into simple chain molecules.
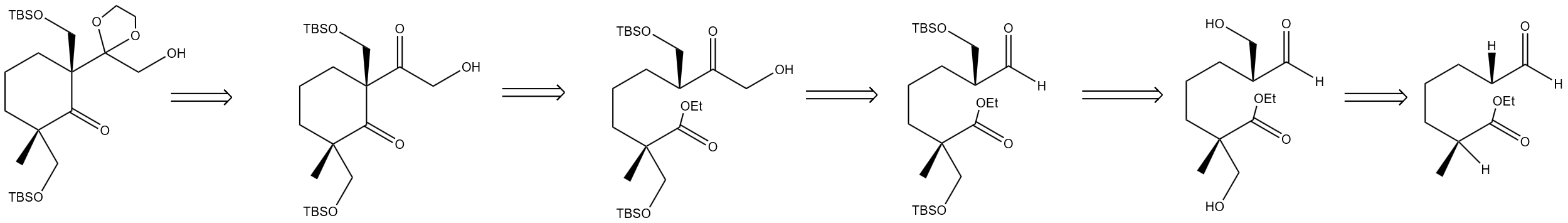
Finally, in figure 10, the molecules with ethyl ester and aldehyde cyclic ketone structures can be reversibly derived as hydroxyl precursors. The hydroxyl precursor is further pushed back and returned to the unsaturated ring structure containing alkene bonds and TMSO protective groups. The unsaturated ring structure continues to be pushed backwards, and is decomposed into a simple cyclic ketene by removing the TMSO protective group and constructing alkene bonds [5].
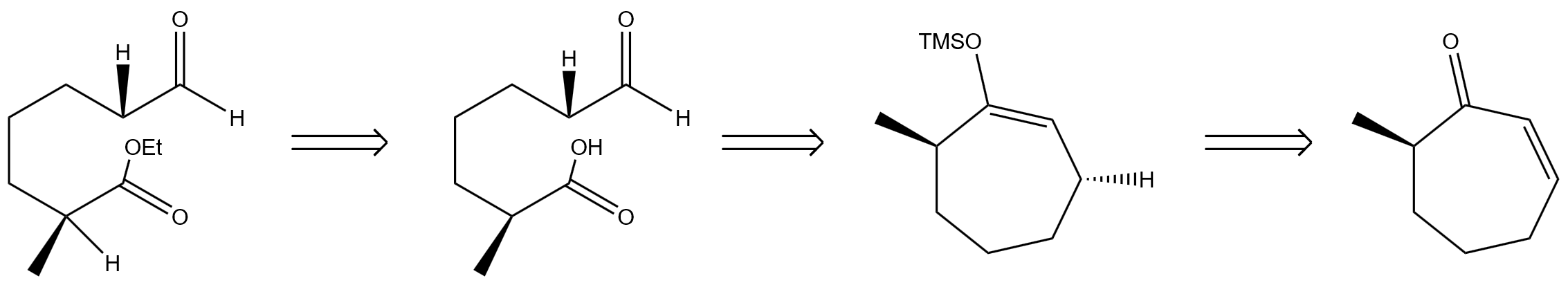
2.2. Synthesis route 1
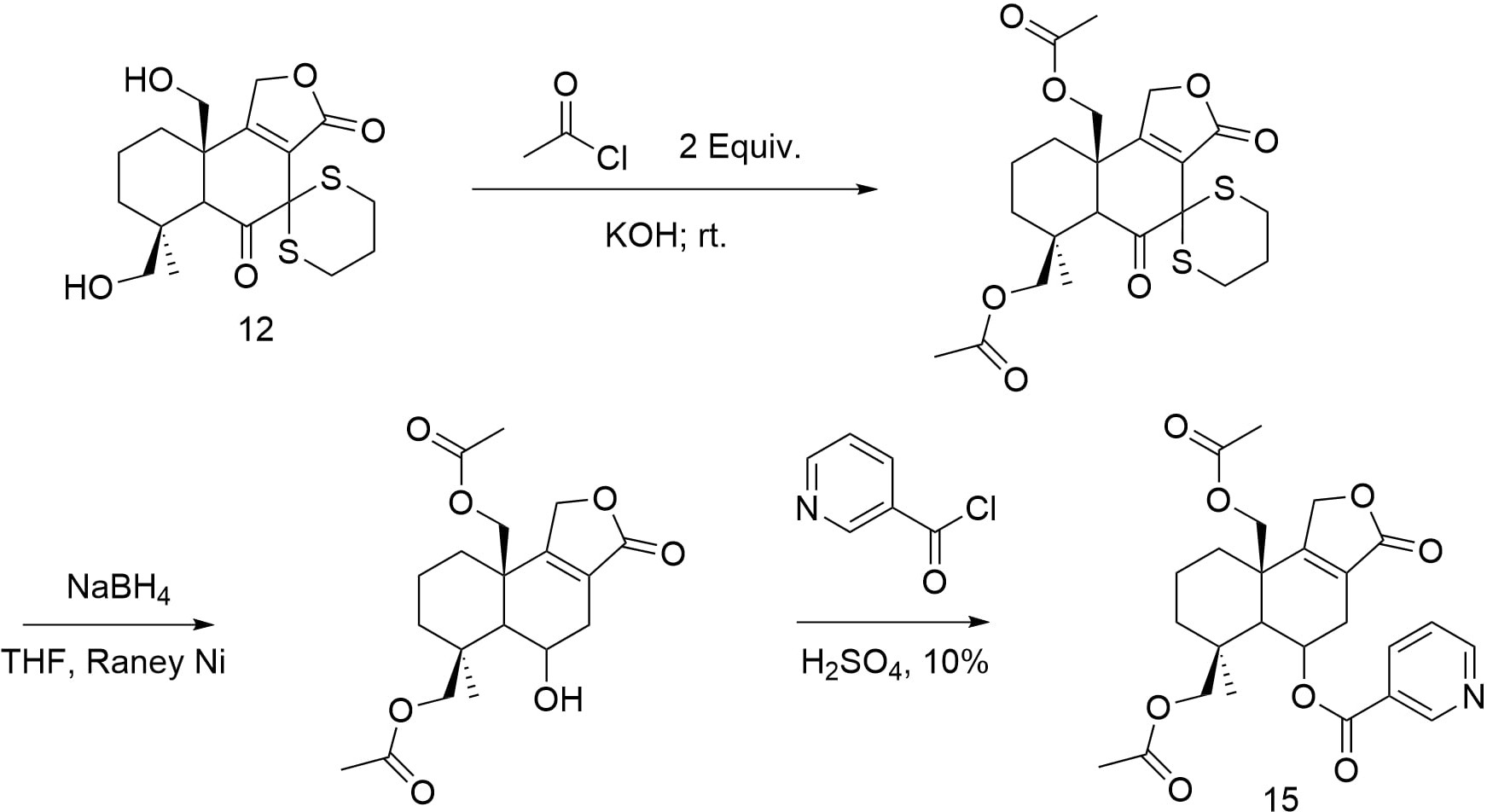
Starting from cycloheptenone derivatives in figure 11, the target ester aldehyde structures were constructed by reacting with Cu(OTf)2, (R,S-S)-L, [CuH(PPh3)]4, Et3N, TMSCl, and HMPA to form TMSO-containing intermediates [5]. The intermediate is then oxidized with ozone, treated with pyridine, then reduced with NaBH4, and then treated with EtOH and H2SO4. Once the ester aldehyde backbone is established, it represents the completion of the synthetic transformation shown.

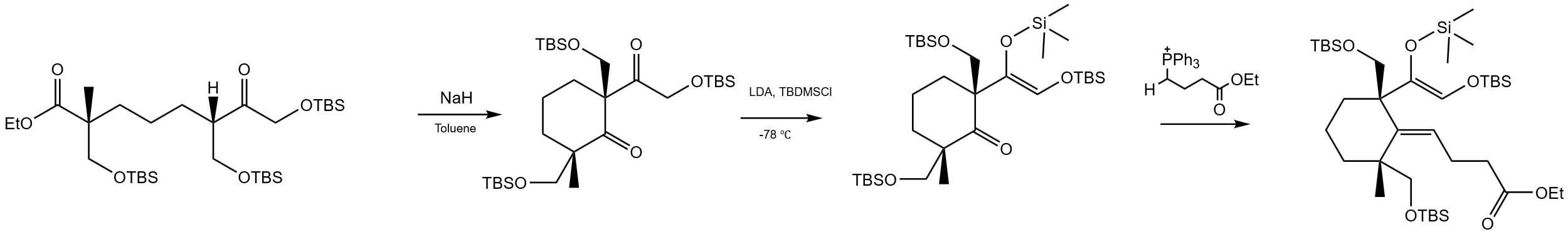
Then starting with the ester-aldehyde compound in figure 12, formaldehyde is first used to react with it under alkaline conditions, introducing two hydroxyl groups by aldol reaction condensation. It then reacts with formaldehyde again to lengthen the carbon chain on the side of the aldehyde carbonyl group [4]. Subsequently, TBSCl and Et3N were used for hydroxyl protection, so as to block the group with TBS to obtain the expected compound.

After that, as it is confirmed in figure 13, treatment with NaH in toluene initiates intramolecular cyclization to form cyclic intermediates containing TBSO and carbonyl groups. Subsequently, it reacts with LDA and TBDMSCl at -78 °C to promote the formation of carbon-carbon double bond and generates an intermediate containing double bonds and protective groups. Then, the Wittig reaction was initiated by the reaction with the phosphorus ylide, and the side chain containing ethyl ester was introduced to obtain a cyclic compound with side chain, which laid the foundation for further synthesis operations.
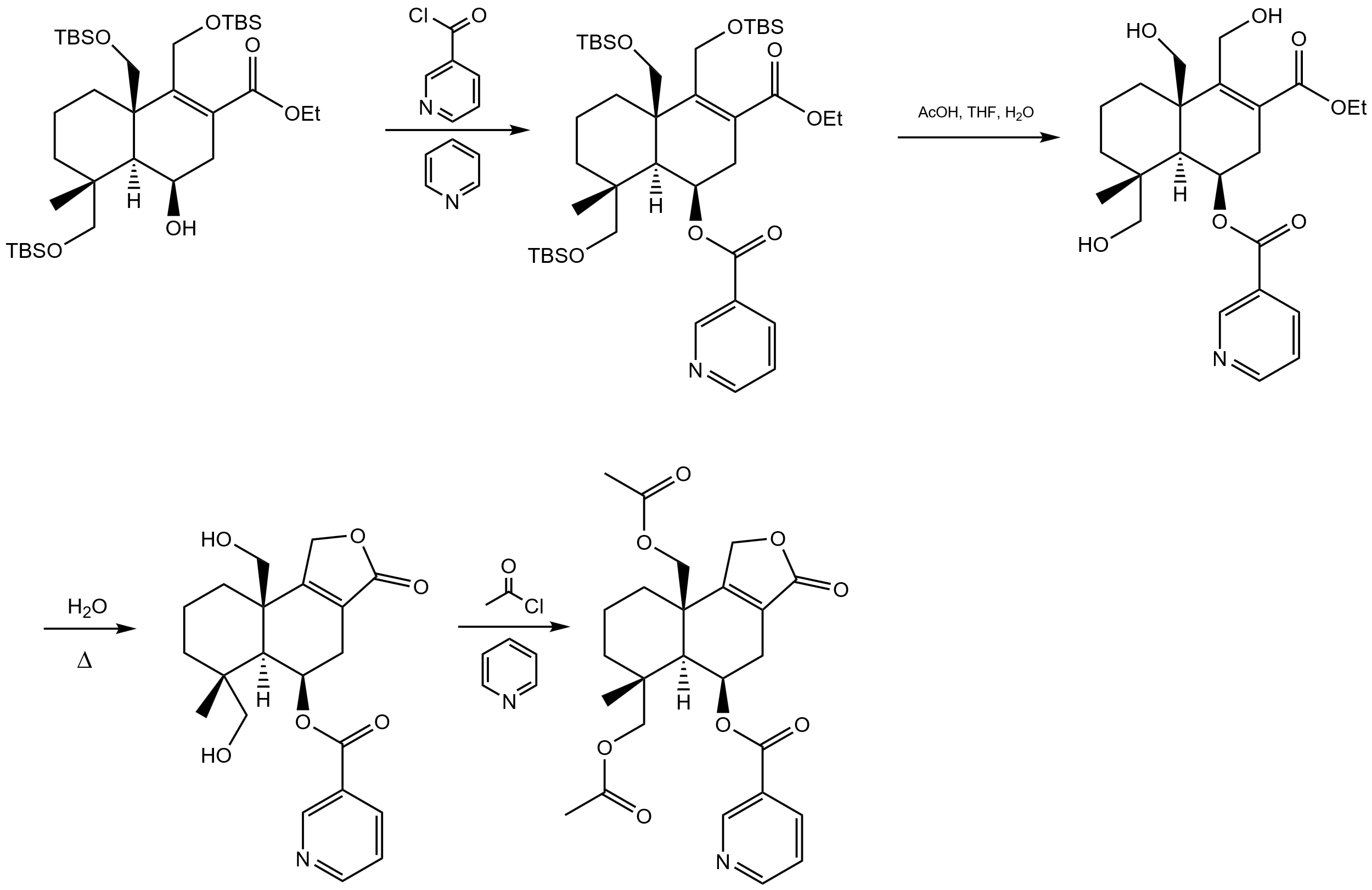
Then, in figure 14, TBAF is used to deprotect the previously protected hydroxyl group to obtain an enol-style structure, and at the same time, according to the tautomerism, the molecule will be transformed into ketones. At this time, the addition product of the anti-markovnikov rules is obtained through the brown hydroboration reaction and addition reaction, and the alkene is changed to alcohol. Then, the main structure of decahydronaphthalene was obtained by aldol condensation and dehydration, and double bonds were formed between carbon atoms 8 and 9 through tautomerism.

Finally, by reacting with pyridine and nicotinoyl chloride, the nicotinic acid building block in figure 15 is reacted with the hydroxyl group generated in the previous step, which reflects the previous use of TBSCl to protect the remaining hydroxyl groups and ensures the selectivity of the reaction. All hydroxyl protection is then removed in preparation for the next reaction [6]. Then, the esterification reaction is used to form the lactone, and then react with acyl chloride and pyridine to obtain the desired product.
Retrosynthesis and synthesis route 1 is a direct synthetic strategy was employed to convert the tricyclic ring into a functionalized cyclopropane derivative in a single chemical operation. This transformation involves the tandem disassembly of one ring of the tricyclic system coupled with the concerted construction of the cyclopropane ring, effectively replacing a ring junction with a cyclopropane bearing several synthetically versatile side chains. This route provides a significant molecular simplification solution. However, there are 4 chiral carbons, which makes some steps could lead to unwilling products in stereochemistry, and making use of protection of functional group several times may be complicated in the experiment.
3. Retrosynthesis and synthesis route 2
3.1. Retrosynthesis route 2
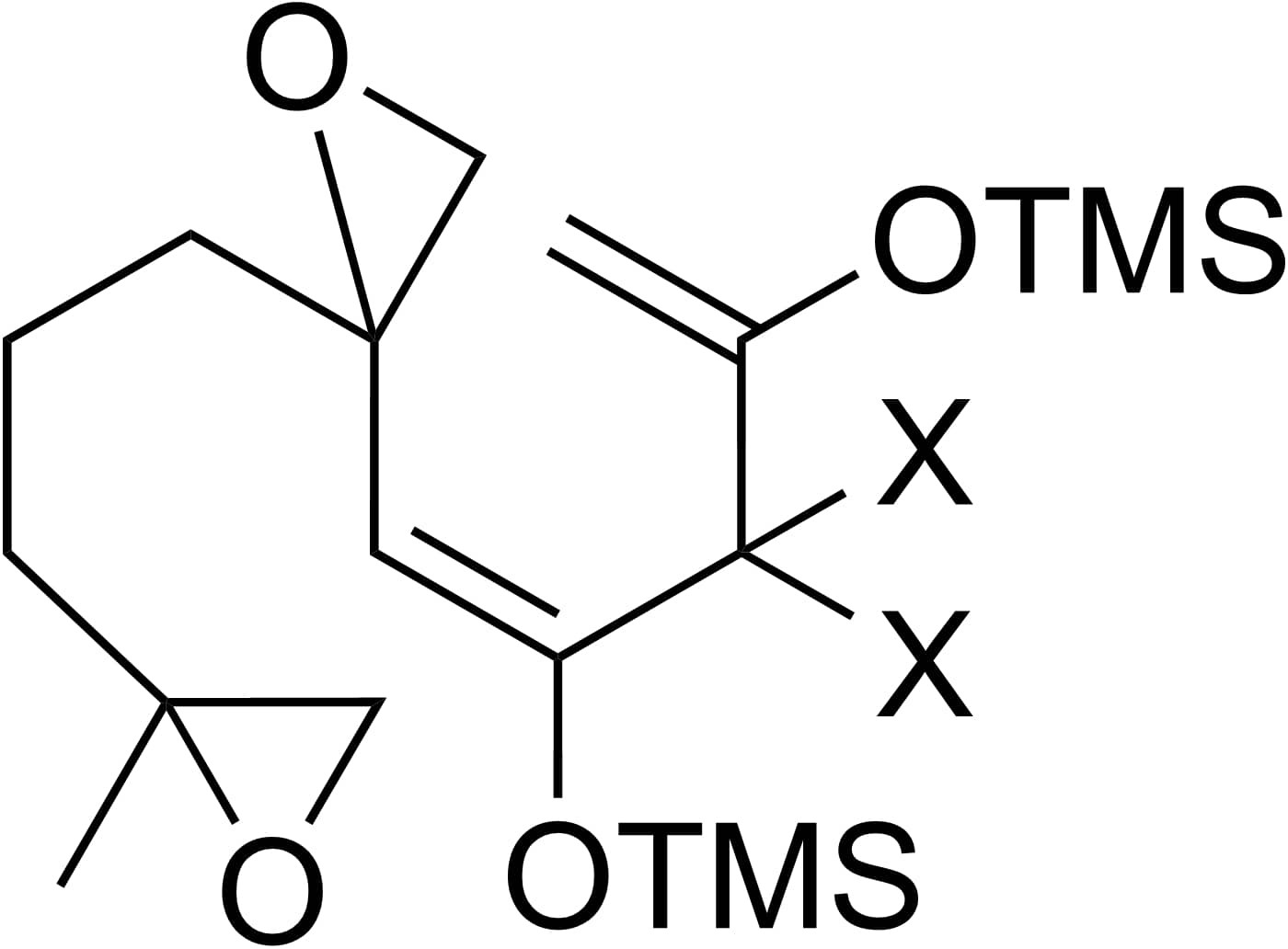
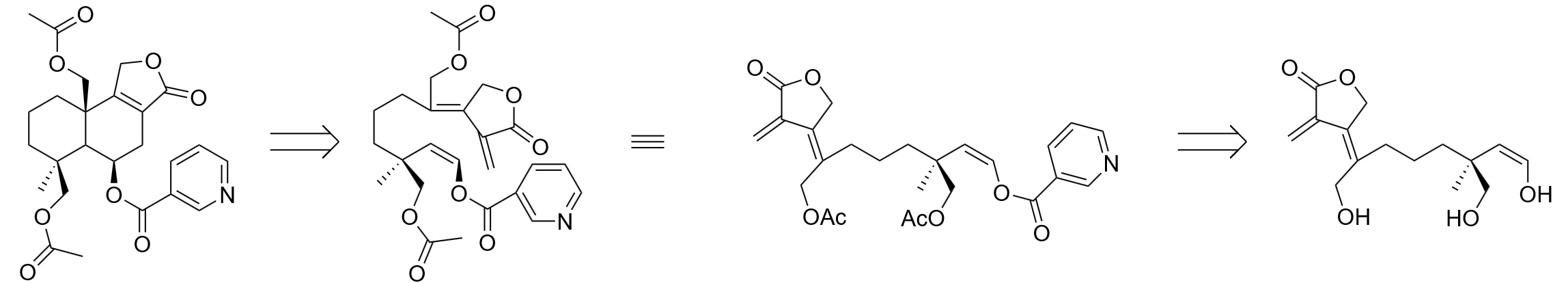
The main structure of Astellolide S is composed of a decalin skeleton and a five-member lactone. To construct the basic decalin skeleton with a specific methylene group (-CH2OH) substituted at the desired position on the decalin skeleton, a unique ring closure scheme incorporating a diepoxide system is provided as shown in figure 16.
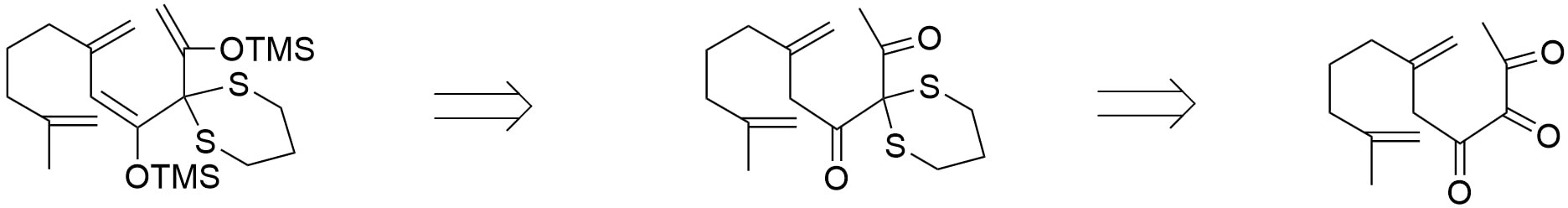
With the preferred di-enol-epoxide system con- structed, the ring closure of the construction of the decalin structure can be completed in one step. To obtain the desired di-enol system from di-ketone, a block in between the two carbonyl groups is re- quired to prevent the di-enol system from being in a conjugated pattern. The blocking can be derived from a vicinal-tri-carbonyl system by converting the middle carbonyl to a dithioether as shown in figure 17. The use of a dithiol to construct the desired molecular block is anticipated to be successful, owing to the pronounced electrophilic nature of the central carbonyl group within the vicinal tricarbonyl system [7]. The vicinal tricarbonyl framework is assembled through the reaction of phosphorus ylide with acyl chloride.
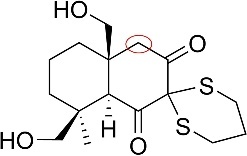
After completion of the ring closure by the di-epoxide system, the resulting 1,4-dienol intermediate tautomerizes back to the corresponding 1,3- dicarbonyl compound. The subsequent synthetic steps focus on constructing the five-membered lactone bearing the upper carbonyl group. Formation of this lactone involves the transformation of the upper carbonyl into a carboxylic acid, which can be accomplished via a palladium-catalyzed carbonylation [8] coupled with the insertion of a methylene hydroxyl group (–CH2OH). Among the three α-positions adjacent to the dicarbonyl system (as shown in figure 18), the top position is the most suitable site for aldehyde insertion due to its electronic and steric effect.
For the preparation of the acyl chloride base, it can be achieved through ozonolysis of a cyclopentene carbonyl chloride proceeding with the conversion of the ketone into olefins through Wittig reaction in figure 19.
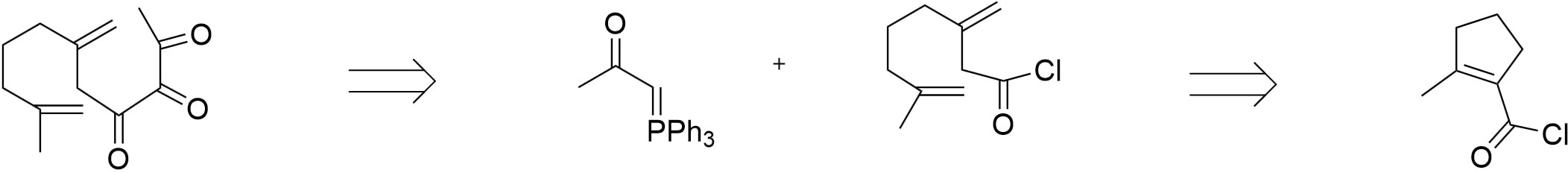
3.2. Synthesis route 2
Starting from cyclopentene carbonyl chloride, the vicinal tricarbonyl system is constructed through sequential ozonolysis followed by reaction with a phosphorus ylide. Prior to the conversion of the phosphorus ylide intermediate into a carbonyl group, the epoxidation of the two olefinic bonds must first be carried out. Once the vicinal tricarbonyl framework is established, subsequent functionalization is performed to introduce a blocking group (dithiol) at the central carbonyl moiety, which is shown in figure 20.
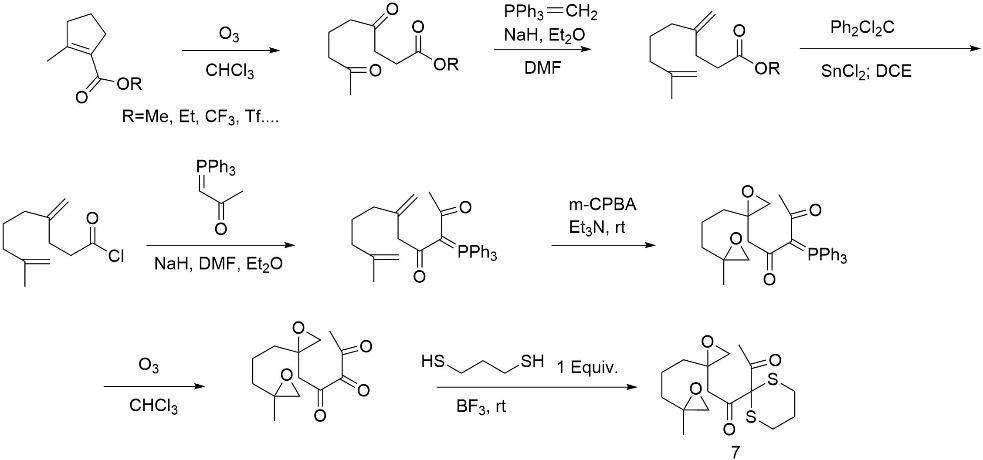
As shown in figure 21, upon preparation of the di-epoxide-dicarbonyl system, enolization facilitates the intramolecular ring closure, thereby constructing the decalin framework characteristic of Astelloide S.
Once the decalin framework has been established, figure 22 outlines the strategy for constructing the lactone moiety. This requires the selective introduction of an aldehyde functionality at the target α-position of the upper carbonyl group. To avoid undesired side reactions during the subsequent palladium-catalyzed carboxylation, the newly formed group must first be protected.
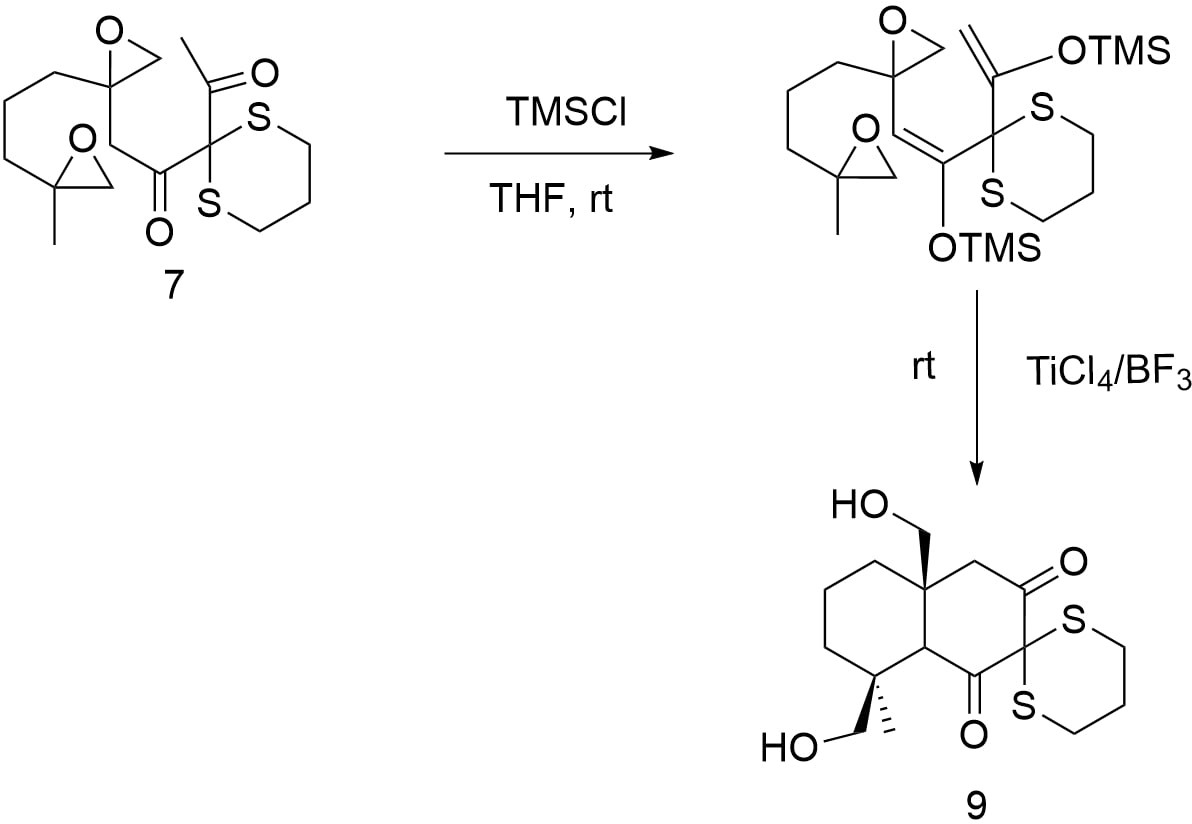
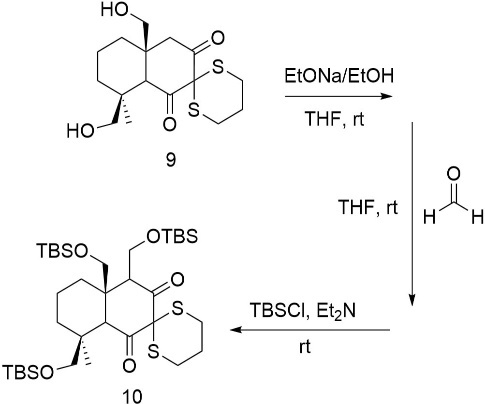
The construction of the five-membered lactone is accomplished through a palladium-catalyzed carbonylation reaction, which involves the insertion of carbon monoxide into the molecular framework. In the process in figure 23, the palladium catalyst facilitates the formation of a new carbon-carbon bond between the activated substrate (typically an alkyl halide or similar leaving group) and carbon monoxide, ultimately yielding a carboxylic acid or ester intermediate. This transformation is crucial for closing the lactone ring and introducing the necessary carbonyl functionality that defines the lactone moiety in the target structure.
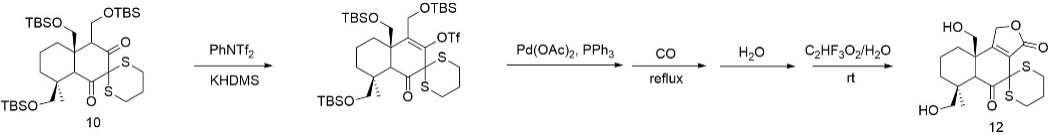
The subsequent steps in figure 24 focus on the installation of the side chains characteristic of astelloide S. These include two methyl acetate moieties introduced at the currently protected hydroxyl group and a nicotinic acid unit attached to the lower carbonyl position. The two methyl acetate groups can be incorporated via a Williamson ether synthesis, employing formyl chloride as the electrophilic component. Following this, the lower carbonyl group is reduced to a hydroxyl using Raney nickel in the presence of sodium borohydride, which simultaneously serves to remove the disulfide protecting group. The newly formed hydroxyl can then undergo a second Williamson ether synthesis to install the nicotinic acid side chain.
As for route 2, By using the delicately designed ring-closure system with epoxides at specific position, the desired decalin-based skeleton of the molecule can be made in one step with desired functional groups substituted in the correct position. Also, the synthesis takes advantage of electrophilic character of the middle carbonyl of the vicinal tri-carbonyl system, to help build the correct and desired ring-closure enol system. But the system contains multi-hydroxyl and carbonyl group potential risking the formation of unwanted byproducts, and the building of the precursor of the ring closure system is complicated.
4. Retrosynthesis and synthesis route 3
4.1. Retrosynthesis route 3
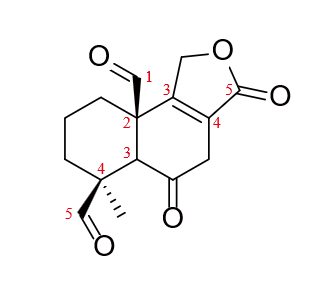
In retrosynthesis route 3, the initial idea was to break the bond by using the double bond of the lactone system. As mentioned in figure 4.
Nevertheless, the feasibility of a Diels-Alder reaction to close the ring is doubtful, because the diene is disubstituted at its terminal. The Z-olefin is also an obstacle to synthesize this precursor molecule.
Another way in figure 25 to break the bonds follows the traditional method of oxidation state analysis. To simplify the analysis, all ester groups except the lactone has been picked out, leaving three hydroxyl groups. Then all three hydroxyl groups are converted into carbonyls for the sake of oxidation state analysis. Two sets of 1, 5 deoxygenation systems are found in this molecule.
Figure 26 shows how we broke the decalin system. To maintain the oxidation state on C-9 while avoiding a triple bond inside the lactone, a halogen atom is added to C-9 in molecule 1. We then broke the bonds between C-9, C-10 and C-4, C-5 and added bonds between C-8, C-9 and C-5, C-10 to obtain molecule 3. It is possible for molecule 3 to build two bonds in one step instead of two Aldol reactions described in Figure 26. To conduct the cascade reaction, a base has to be used in order to remove the proton on C-4. However, this can be problematic because the proton on C-7 is more acidic than the one on C-4. A negative charge would occur on C-4, which could not give birth to our target molecule. To get rid of this side reaction, we converted the carbonyl on C-7 into a hydroxyl group and make further protection on molecule 4 in figure 27.
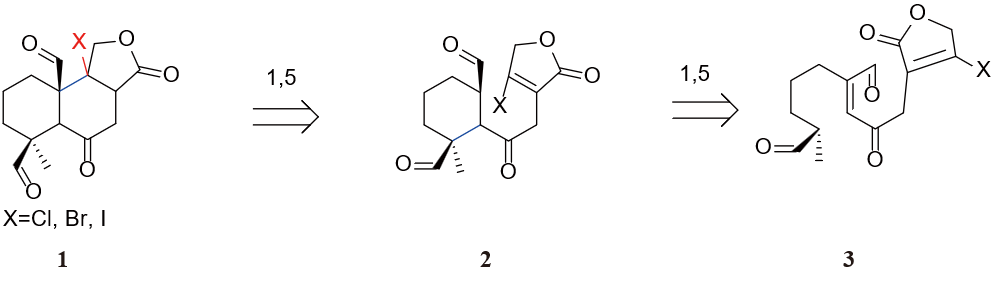
For molecule 4 in figure 28, the middle carbonyl has formed a 1, 3 deoxygenation pattern with the middle double bond. Thus, we chose to cut the middle double bond, resulting in two small molecules.
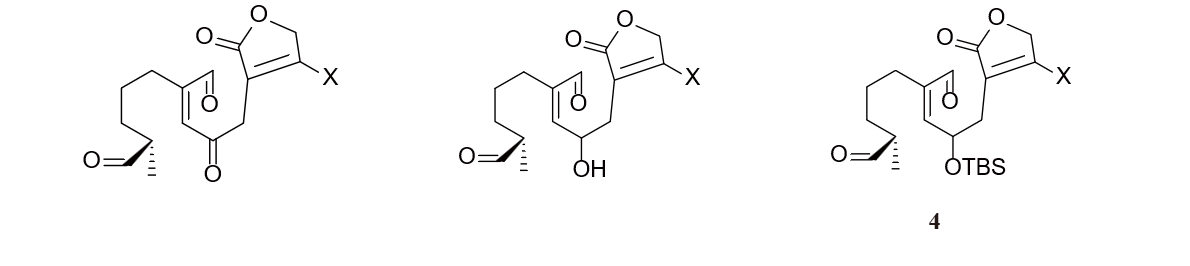
Figure 29 and 30 show the strategy we took in cutting molecule 5 and 6. Noticing that dialdehyde 5 could be viewed as α-position substituted aldehyde, we chose to cut the bond between the α and β position, resulting in two synthesis equiavalents enolate and carbocation. The retrosynthesis analysis of molecule 6 involves an interconversion of bromine to enol which is equilibrium with ketone, followed by converting a 1,2 dioxigenation pattern (molecule 7) back to a double bond (molecule 8) so as to avoid substitution reaction on sp2 hybridized carbon. Finally diving the alkene part from the lactone part produces molecule 9 and 10 in figure 31 as two raw materials.


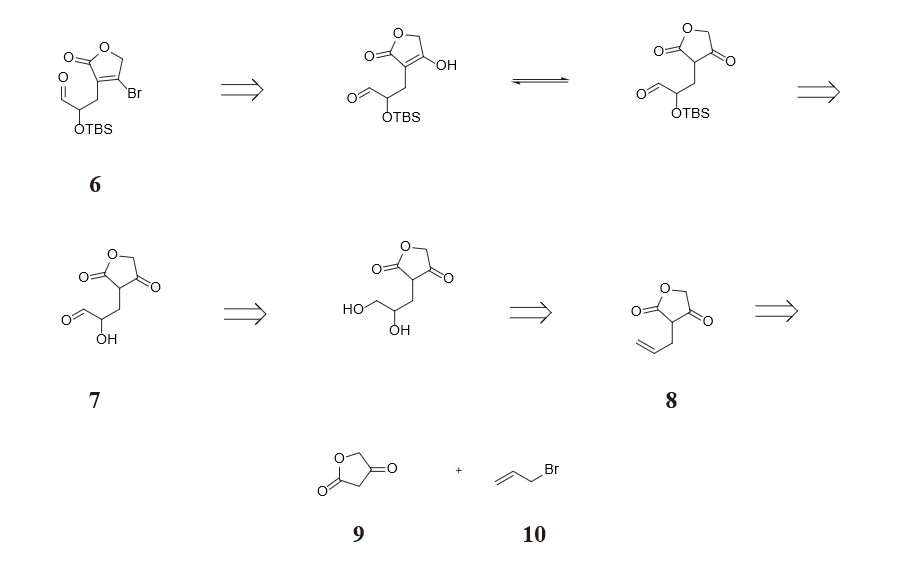
4.2. Synthesis route 3
Our synthesis route 3 starts with the construction of molecule 16, which is in figure 32. 5-iodo-2-methylpent-1-ene 11 is treated with borane in tetrahydrofuran. The resulting primary alcohol 12 is then oxidized by pyridinium chlorochromate in dichloromethane so that an aldeheyde 13 is produced. An additional step of ethylene glycol protection on the carbonyl is necessary to avoid side reactions in construcuting the decalin system, of which the reason will be explained later. Molecule 14 is treated with pseudoephedrine to make α-substituted 6-(1,3-dioxolan-2-yl) heptanoyl iodide 15, followed by reduction with LiAlH(OEt)3 to convert carboxylic acid iodide to the aldehyde 16.
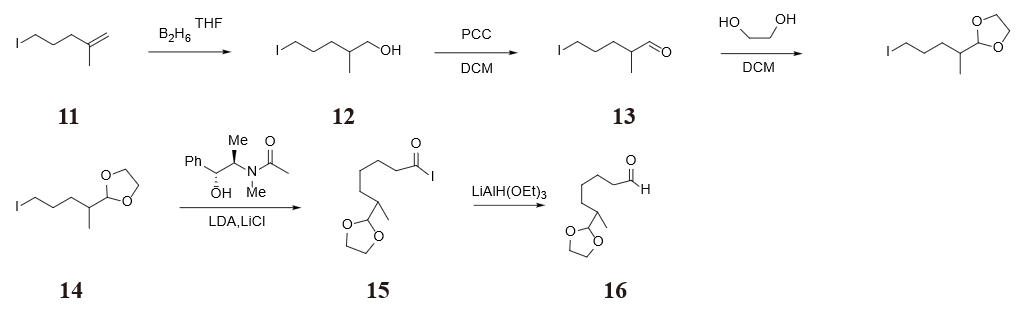
Myers discouvered a method to produce α-substituted carbonyls through pseudoephedrine and alkyl halide or its derivatives in 1994 [9]. By using different reagents in the second step carbopxylic acid halide could be convereted to carboxylic acid, aldeheyde or primary alcohol. One adavantege of Myers reaction is the formation of a chiral center, as it is shown in figure 33. An other advantege lies in the high yield in both of the two steps: around 90% in substitution and around 77% in reducing carboxylic acid chloride to an aldehyde.

To construct molecule 6, furan-2,4(3H,5H)-dione 17 is treated with sodium ethoxide and 3-bromoprop-1-ene. The generated 3-allylfuran-2,4(3H,5H)-dione 18 is oxidized by osmium tetroxide to give a diol 19. Then the primary alcohol is selectively oxidized to a carbonyl by TEMPO, while the secondary alcohol is protected by using TBSCl [10].
Hydrogen bromide and heating is applied at last to react with the ketone carbonyl on the lactone to generate molecule 6 in figure 34.
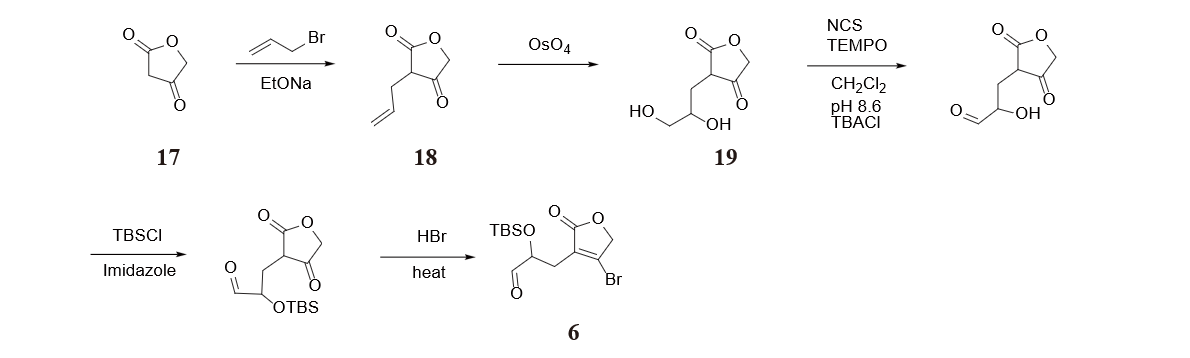
Figure 35 shows that after obtaninig molecule 6 and molecule 16, LDA is applied to connect these two molecules. Treat molecule 20 with an acid to convert the acetal protaection back to a carbonyl (molecule 21), which serves as a precursor of the following ring closure reaction. A proposed mechanism of ring closure reaction and its next step are also displayed in Figure 35. After treating molecule 21 with LDA, the most acidic proton has been removed. The resulting carbanion attacks the double bond, which attacks the bromine substituted carbon in the lactone, causing the bromine to leave. The final step include the use of TBAF to take off the TBS group, producing molecule 22 as a precursor of esterification.
To avoid confusing three ester groups, we decided to first make an ester group at C-6 in molecule 22, followed by a reduction with sodium borohydride, as is shown in figure 35. By treating the resulting molecule with two equivalents of acetic acid chloride can we obtain the target molecule Astellolide S.

It is worth mentioning that molecule 6 in retrosynthesis analysis and molecule 16 in synthesis is different in one functional group, but they serve the same purpose. We designed an acetal protection in order to avoid a predominant six-member ring stable byproduct 3-methylcyclohex-1-ene-1-carbaldehyde. Figure 36 shows the proposed mechanism of how this byproduct form, together with three other possible by products.

In figure 37, Molecule 23 and 24 are possible dimers of molecule 6 instead of a self-ring closure that gives the six-member ring byproduct. Molecule 25 originates from LDA removing the lower α-proton instead of the above one and reacting with molecule 6 at the wrong position. However, applying an acetal protection could eradicate the risk of self ring closure and totally avoid the formation of a stable byproduct. Though the formation of molecule 23, 24, and 25 are still possible, their yield are believed to be low. Therefore, the protection we made makes sense and could absolutely increase the yield of the target molecule.

Route 3 provides a rather simple way to synthesize the target molecule. All reagents are common ones so that the reactions are easy to conduct in labs. In addition, by special designing of molecule, a byproduct which causes much frustration is avoided. The precursor of ring closure is easily synthesized. Nevertheless, the reaction still produces other kinds of byproducts, they by no means can be avoided in this route.
5. Conclusion
All in all, the stereochemical selectivity of all three routes is difficult to guarantee, and this issue still needs to be further studied. Given the unique and rich reactive functional groups (such as rare nicotinic acid building block and γ-lactones) and the significant antimicrobial activity already demonstrated by its family members, future research can focus on developing efficient and stereoselective full synthesis routes, such as optimizing existing strategies or exploring bioinspired synthesis, to obtain sufficient samples. This will provide a solid foundation for in-depth study of its biological activity mechanisms, structure-activity relationships, and the development of its potential as novel antimicrobials or other therapeutic agents.
References
[1]. Dai G, Sun J, Peng X, et al. Astellolides R–W, Drimane-Type Sesquiterpenoids from an Aspergillus parasiticus Strain Associated with an Isopod [J]. Journal of Natural Products, 2023, 86(7): 1746-53.
[2]. Aminudin N I, Ridzuan M, Susanti D, et al. Biotransformation of sesquiterpenoids: a recent insight [J]. Journal of Asian Natural Products Research, 2021, 24(2): 103-45.
[3]. Einhorn J, Einhorn, C., Ratajczak, F., & Pierre, J.-L. Efficient and Highly Selective Oxidation of Primary Alcohols to Aldehydes by N-Chlorosuccinimide Mediated by Oxoammonium Salts. [J]. The Journal of Organic Chemistry, 1996: 61(21):7452–4.
[4]. Cao H, Liu Y, Li Y, et al. N-heterocyclic carbene promoted one-pot synthesis of 1, 2-diols and ɑ-Hydroxyketones from simple aldehydes [J]. Molecular Catalysis, 2024, 564.
[5]. Andringa R L H, Marinus N, Bunt D V, et al. Total synthesis of dissectol A, using an enediolate-based Tsuji–Trost reaction [J]. Chemical Science, 2024, 15(27): 10541-6.
[6]. Sugimoto K, Kobayashi Y, Hori A, et al. Syntheses of aza-analogues of macrosphelides via RCM strategy and their biological evaluation [J]. Tetrahedron, 2011, 67(40): 7681-5.
[7]. PARR H H W A J. The Chemistry of Vicinal Tricarbonyls and Related Systems [J]. ACCOUNTS OF CHEMICAL RESEARCH, 2004: 37: 687-701.
[8]. Nicolaou K C, Baran P S, Zhong Y L, et al. Total Synthesis of the CP-Molecules (CP-263, 114 and CP-225, 917, Phomoidrides B and A). 2. Model Studies for the Construction of Key Structural Elements and First-Generation Strategy [J]. Journal of the American Chemical Society, 2002, 124(10): 2190-201.
[9]. Myers A G, Yang, B. H., Chen, H., & Gleason, J. L. Use of Pseudoephedrine as a Practical Chiral Auxiliary for Asymmetric Synthesis. [J]. Journal of the American Chemical Society, 1994: 116(20):9361–2.
[10]. Shinohara Y, Takahashi S, Osada H, et al. Identification of a novel sesquiterpene biosynthetic machinery involved in astellolide biosynthesis [J]. Sci Rep-Uk, 2016, 6(1).
Cite this article
Chen,G.;Wang,Z.;Shi,Z. (2025). Retrosynthetic Analysis and Design for the Total Synthesis of Astellolide S. Applied and Computational Engineering,200,7-24.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of CONF-MCEE 2026 Symposium: Advances in Sustainable Aviation and Aerospace Vehicle Automation
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Dai G, Sun J, Peng X, et al. Astellolides R–W, Drimane-Type Sesquiterpenoids from an Aspergillus parasiticus Strain Associated with an Isopod [J]. Journal of Natural Products, 2023, 86(7): 1746-53.
[2]. Aminudin N I, Ridzuan M, Susanti D, et al. Biotransformation of sesquiterpenoids: a recent insight [J]. Journal of Asian Natural Products Research, 2021, 24(2): 103-45.
[3]. Einhorn J, Einhorn, C., Ratajczak, F., & Pierre, J.-L. Efficient and Highly Selective Oxidation of Primary Alcohols to Aldehydes by N-Chlorosuccinimide Mediated by Oxoammonium Salts. [J]. The Journal of Organic Chemistry, 1996: 61(21):7452–4.
[4]. Cao H, Liu Y, Li Y, et al. N-heterocyclic carbene promoted one-pot synthesis of 1, 2-diols and ɑ-Hydroxyketones from simple aldehydes [J]. Molecular Catalysis, 2024, 564.
[5]. Andringa R L H, Marinus N, Bunt D V, et al. Total synthesis of dissectol A, using an enediolate-based Tsuji–Trost reaction [J]. Chemical Science, 2024, 15(27): 10541-6.
[6]. Sugimoto K, Kobayashi Y, Hori A, et al. Syntheses of aza-analogues of macrosphelides via RCM strategy and their biological evaluation [J]. Tetrahedron, 2011, 67(40): 7681-5.
[7]. PARR H H W A J. The Chemistry of Vicinal Tricarbonyls and Related Systems [J]. ACCOUNTS OF CHEMICAL RESEARCH, 2004: 37: 687-701.
[8]. Nicolaou K C, Baran P S, Zhong Y L, et al. Total Synthesis of the CP-Molecules (CP-263, 114 and CP-225, 917, Phomoidrides B and A). 2. Model Studies for the Construction of Key Structural Elements and First-Generation Strategy [J]. Journal of the American Chemical Society, 2002, 124(10): 2190-201.
[9]. Myers A G, Yang, B. H., Chen, H., & Gleason, J. L. Use of Pseudoephedrine as a Practical Chiral Auxiliary for Asymmetric Synthesis. [J]. Journal of the American Chemical Society, 1994: 116(20):9361–2.
[10]. Shinohara Y, Takahashi S, Osada H, et al. Identification of a novel sesquiterpene biosynthetic machinery involved in astellolide biosynthesis [J]. Sci Rep-Uk, 2016, 6(1).