







1 Introduction
The shape or conformation of a protein fold depends directly on its linear amino acid sequence [1]. The folded structure of a protein is stabilized by thousands of noncovalent bonds between amino acids [1]. The way in which the exposed surface of a protein interacts with other molecules determines the function of the protein [1]. In order to understand the 3D protein structure, X-ray crystallography, electron crystallography and nuclear magnetic resonance techniques are mainly used in addition to the computational method, i.e., prediction from amino acid sequence [2]. Traditional experimental methods usually require expensive equipment, time and effort. On the other hand, the prediction of 3D protein structures ranges from ab-initio methods based entirely on physicochemical principles, homology methods based mainly on information available in sequence and structural databases and thread or fold recognition [3].
In CASP14 [4], AlphaFold2 achieved excellent results. In the absence of similar protein structures, it can periodically predict protein structures with atomic accuracy [5]. Using primary amino acid sequences and homologous aligned sequence profiles as input, the AlphaFold2 network directly predicts the 3D coordinates of all heavy atoms of a given protein with an equivariant attention architecture [5]. However, the prediction of protein structure by AF2 is not involved in ligands or small molecule drugs. AF2complex predicts the structures of multimeric protein complexes [6]. In this study, AlphaFold2 was used to attempt to predict the structure of a protein in complex with a covalent inhibitor.
Receptor tyrosine kinases (RTKs) are transmembrane proteins that regulate normal cellular processes by transducing signals from the extracellular environment to the cytoplasm and nucleus [7]. The increase in RTK expression or activation is directly related to the pathogenesis of various human cancers [7]. The ErbB family of receptor tyrosine kinase (RTKs) has four members: epidermal growth factor receptor (EGFR), ErbB2(HER2), ErbB3(HER3), and ErbB4(HER4) [8]. After the receptor binds to the ligand, conformational changes will occur, forming homodimers or heterodimers, and finally initiating downstream signaling events [8]. Although ErbB2 does not directly bind any ErbB ligand, ErbB2 is the preferred heterodimerization partner for all ErbB proteins [9]. Overexpression or amplification of HER2 is associated with the malignant degree and poor prognosis of breast cancer [10]. HER2 levels are elevated in 20% of breast tumors [8]. One of the most important pathway activated downstream of HER2 in cancer is that HER2 signals PI3K/Akt together with HER3 [8].
This research [11] identified covalent tyrosine kinase inhibitors that efficiently inhibit HER2 exon 20 mutants while retaining WT EGFR, which reduced tumor cell survival and in vitro proliferation and inhibited downstream HER2 signaling. X-Ray crystal structure data of HER2 TKI bound to HER2 as a ligand are stored in the Protein Data Bank [12] under code 7PCD [13]. In this study, HER2YVMA inhibitor will be used as an example to observe the conformational changes of HER2 protein and try to reveal more about the mechanism of drug discovery.
2 Methods
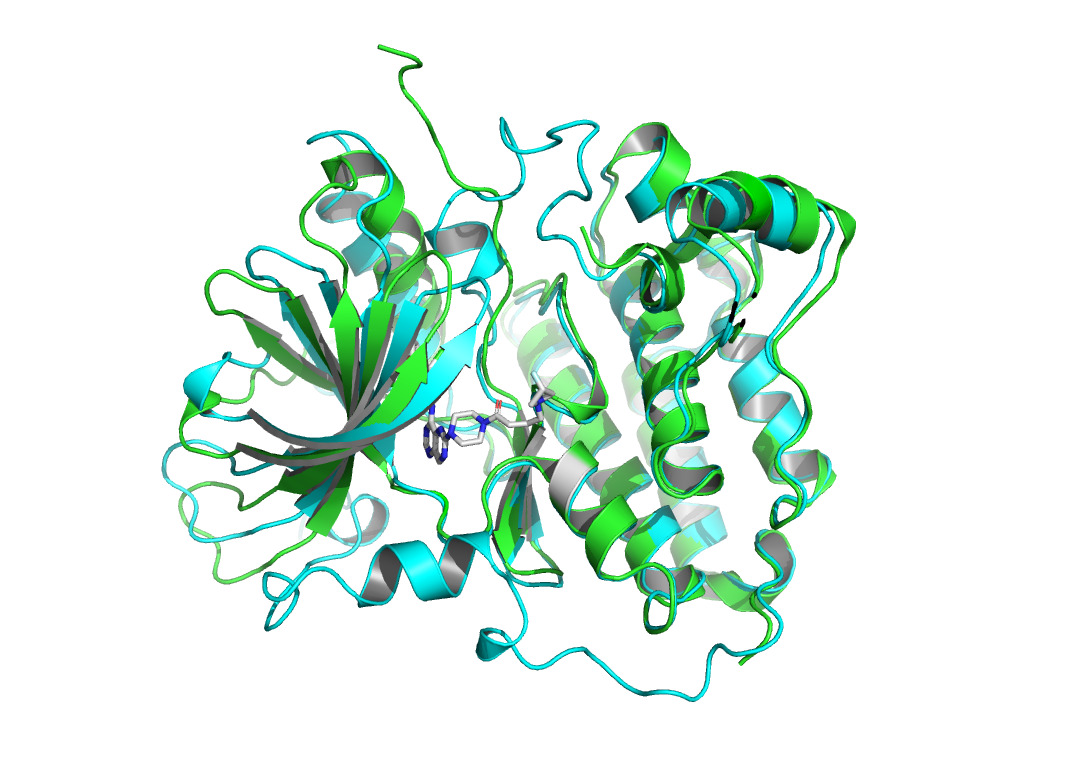
Given that AlphaFold2’s predictions are only for proteins without ligand involvement, this study focuses on protein structure predictions that form complexes with inhibitors. This work observed the structural differences and investigated the conformational changes of HER2 protein after binding a ligand. It collected the protein structure after binding from the Protein Data Bank [12]. 7PCD [13] was selected, which is HER2 in complex with a covalent inhibitor. PyMOL [14] was used to observe the conformational changes of the protein before and after binding in figure 1.
|
Figure 1. X-ray crystal structure of HER2 before and after binding to the inhibitor. AlphaFold2 predicted HER2 pre-binding structure is shown in green. The structure of HER2 after binding to the inhibitor (represented as sticks in gray and atom colors) is shown in cyan. |
|
Figure 2. The visualization of interacting chain A shown by PLIP [15]. |
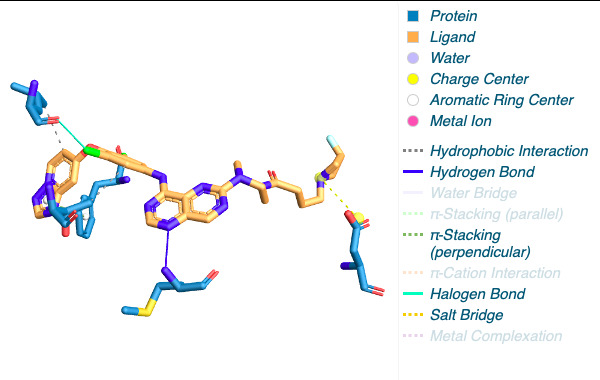
By using the Protein-Ligand Interaction Profiler [15], table 1 demonstrates the non-covalent interactions between HER2 Protein A Chain and its covalent inhibitor. Figure 2 shows the visualization of interacting chain A shown by the PLIP [15]. In figure 3, interactions are labeled by light magentas. Protein structural changes caused by the protein-protein interaction can be found.
Table 1. The non-covalent interactions between HER2 Protein A Chain and its covalent inhibitor shown by PLIP [15]. |
Index |
Interaction |
Residue |
AA |
1 |
Hydrophobic Interactions |
796A |
LEU |
2 |
Hydrophobic Interactions |
798A |
THR |
3 |
Hydrogen Bonds |
801A |
MET |
4 |
π-Stacking |
864A |
PHE |
5 |
Halogen Bonds |
796A |
LEU |
6 |
Salt Bridges |
808A |
ASP |
3 Results and Discussion
|
Figure 3. The interactions are labeled by light magentas using PyMOL [14]. |
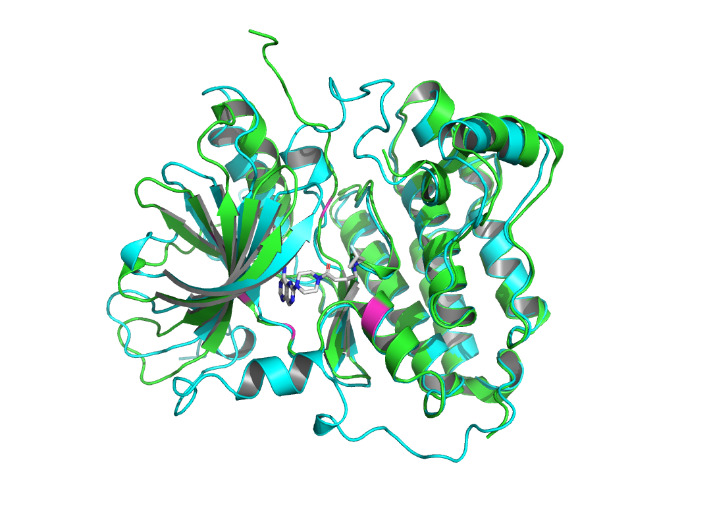
The non-covalent bonds formed by the binding of proteins and ligands have energy and can cause local conformational changes in proteins. The structure difference of protein before and after binding with ligand may be due to the error of protein structure prediction or the conformational change of protein. The binding sites identified by PLIP [15] are labeled in figure 3. After comparing the structure of HER2 protein in the free state with the structure after ligand binding, some shifts near the binding sites were found. This further verifies the formation of non-covalent bonds in the complex. At the same time, this also reflects the accuracy of AF2 in predicting the structure of ligand-binding HER2 to a certain extent.
|
Figure 4. Residue 796A labeled by light magentas using PyMOL [14]. |
|
Figure 5. Residue 798A labeled by light magentas using PyMOL [14]. |
|
Figure 6. Residue 801A labeled by light magentas using PyMOL [14]. |
|
Figure 7. Residue 864A labeled by light magentas using PyMOL [14]. |
|
Figure 8. Residue 808A labeled by light magentas using PyMOL [14]. |
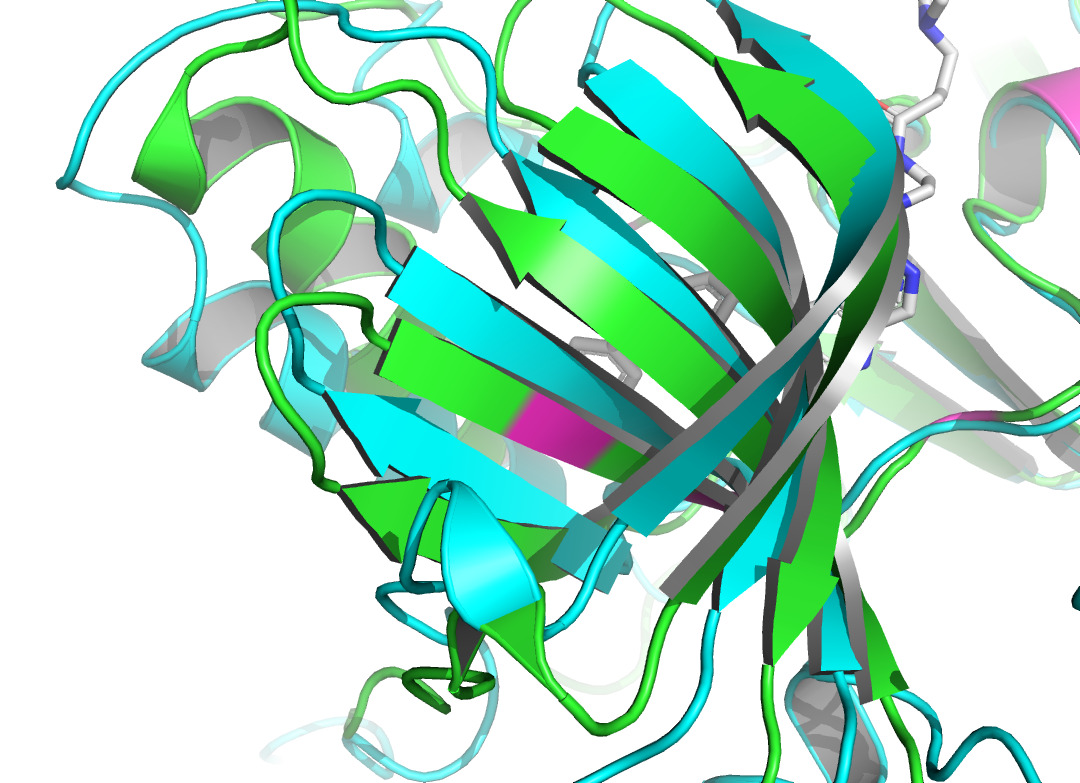
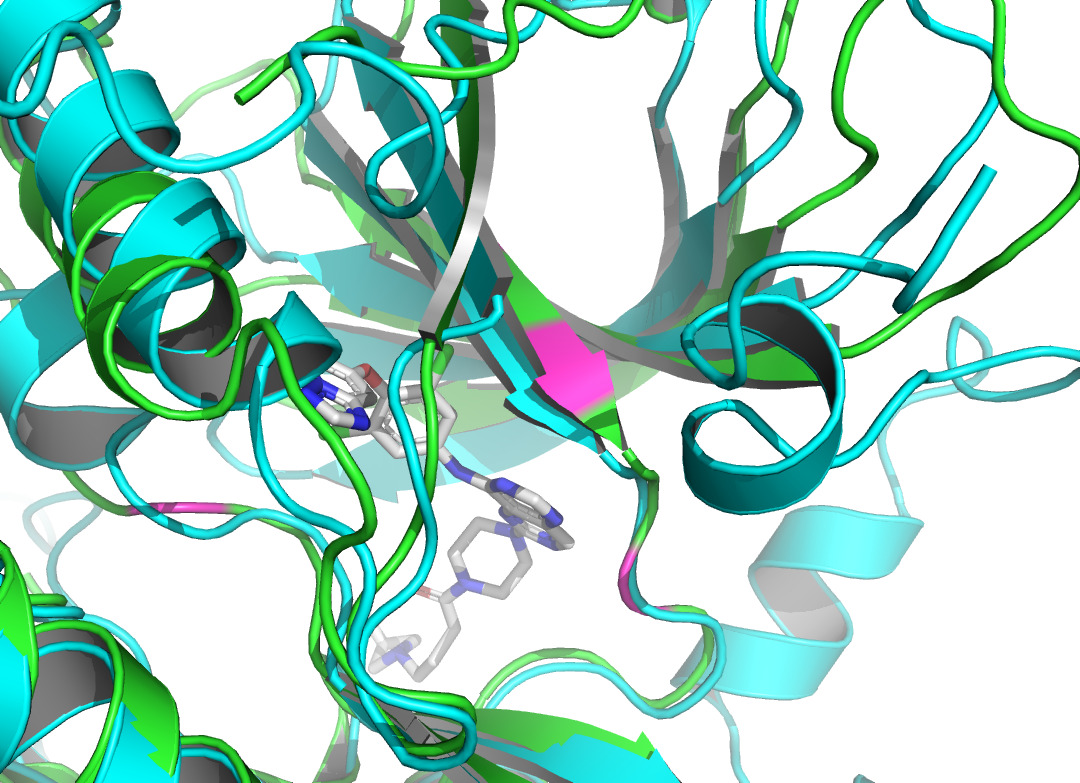
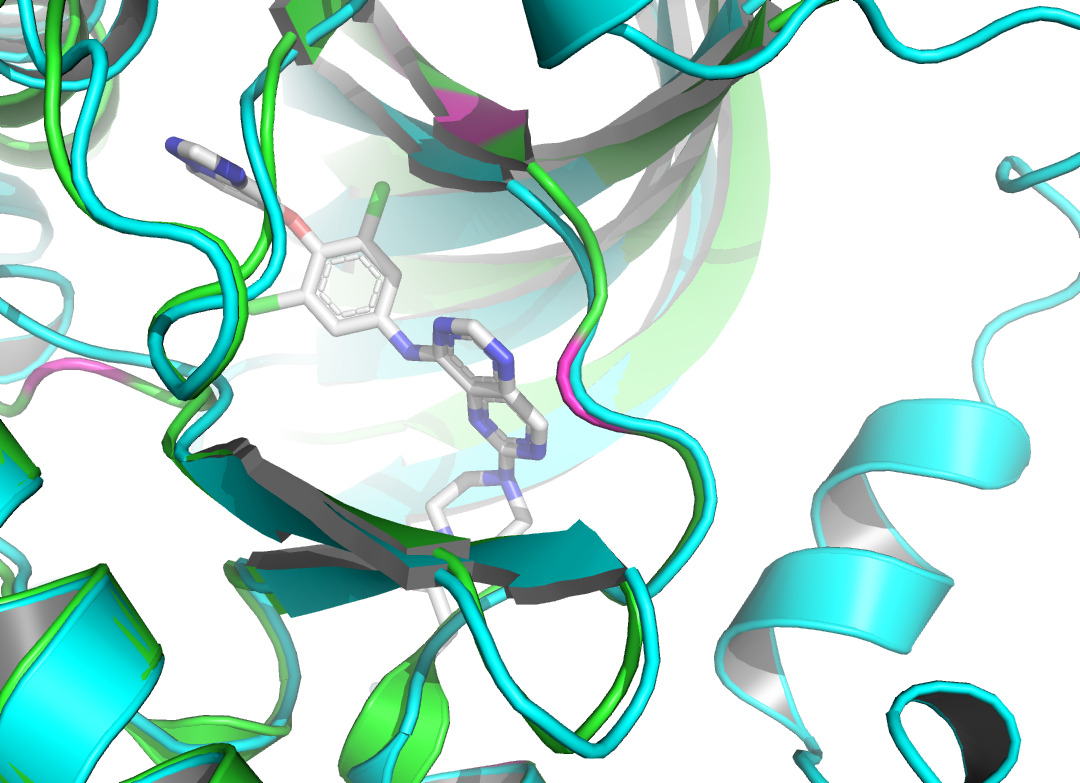
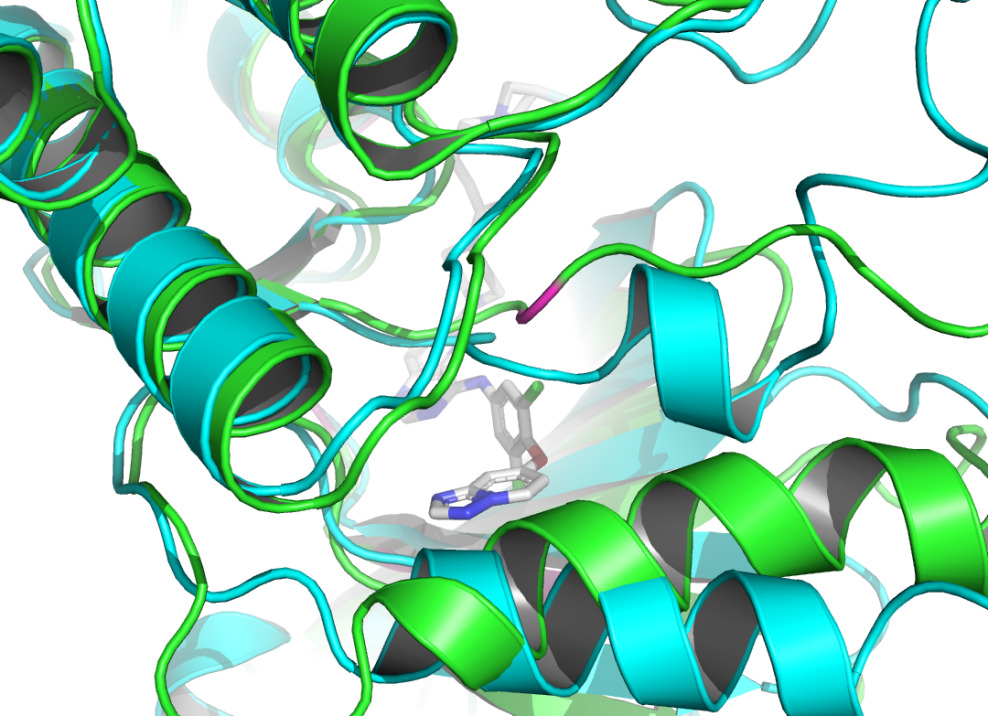
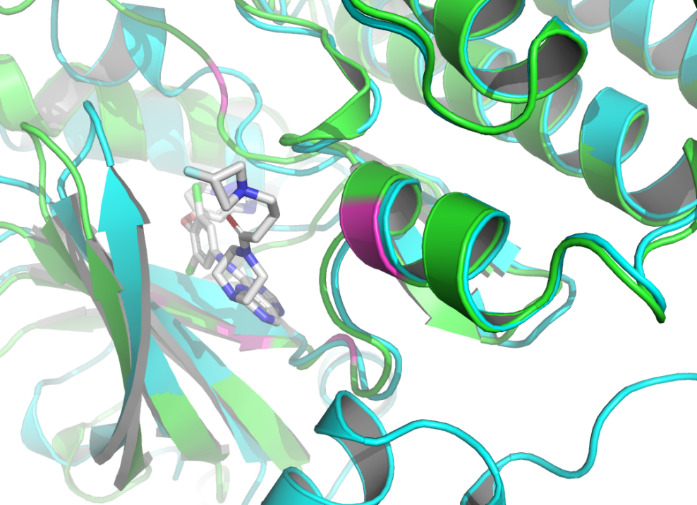
Residue LEU at position 796, located in the A chain of HER2 protein, forms two non- covalent bonds with the HER2 TKI, which are hydrophobic interaction and halogen bond. Residue THR at position 798A also produces a hydrophobic interaction with the ligand. According to figure 4 and figure 5, they both shifted upon ligand binding, probably because of hydrophobic forces. Residue LEU of 801A forms a hydrogen bond with the ligand, and the local secondary structure at this position is not altered according to figure 6. The main reasons may be due to the small force provided by hydrogen bonding compared with hydrophobic interaction, the loose structure of the protein at this site, and the prediction problem. The fourth binding site is at 864A, and a π-stacking is formed at this position. Figure 7 shows that there is a conformational change in this binding site, even though the protein structure at this site is loose. A salt bridge is located at 808A, see figure 8. The local secondary structure of the site was not altered. The possible reason is that the salt bridge exerts relatively weak forces.
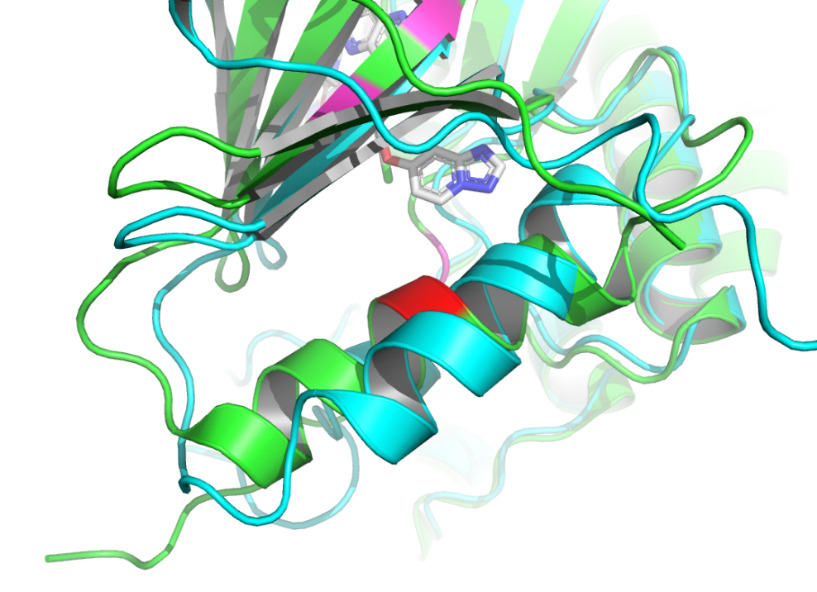
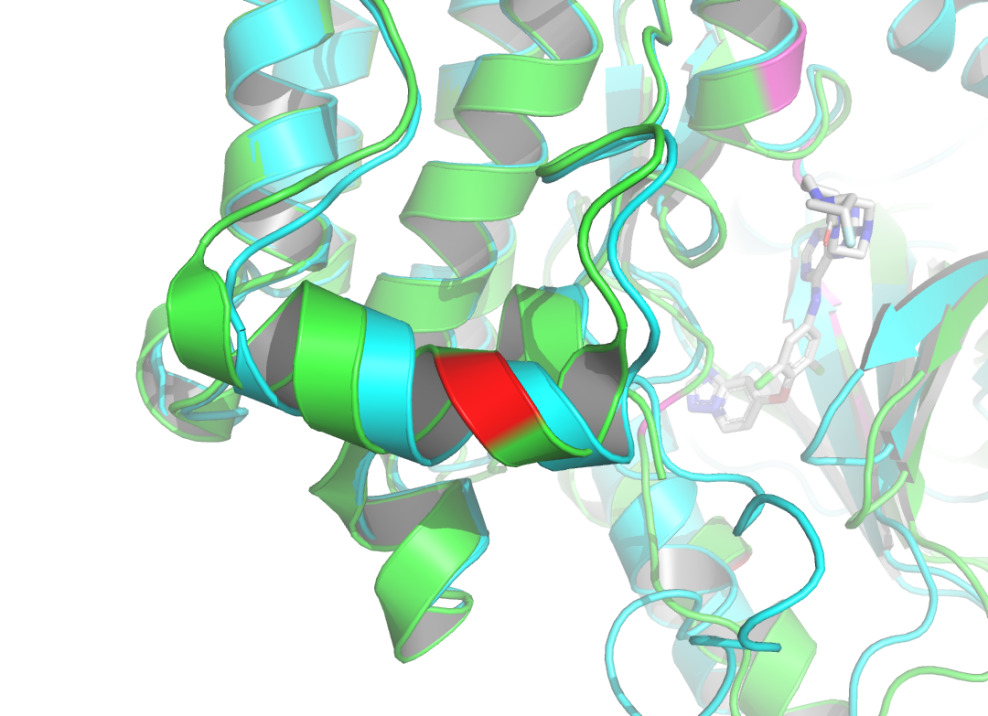
Docking of proteins with ligands is a dynamic binding process due to the influence of the intracellular environment, so there may be potential binding pose and conformational changes that are not captured. In addition to the known binding sites, some other sites of the complex in this study have more structural changes, so it is possible that more binding sites could be identified. According to figure 9 and figure 10, the binding site may be close to 768A and 930A, because the conformational change of this site is obvious. As the prediction of binding sites holds significant importance in drug development, numerous advanced binding site prediction techniques will be explored in depth in future studies.
|
Figure 9. Residue 768A labeled by red using PyMOL [14]. |
|
Figure 10. Residue 930A labeled by red using PyMOL [14]. |
4 Conclusions
Understanding the conformation of drug molecules binding to target proteins is essential for the design and modification of ligand drugs. Determining the binding site of a protein to a ligand or a known small-molecule drug is useful for practical applications, such as the development of HER2-targeting drugs. By comparing the predicted monomer structure generated by AlphaFold2 with the protein structure upon ligand binding, it can indicate some new potential binding sites. This study found two potential binding sites through the observation of conformational changes in HER2 when it interacts with a covalent inhibitor. These sites can provide clues for pharmaceutical chemists in the development of ligand drugs.
References
[1]. O’Connor C M, Adams J U and Fairman J E 2010 Essentials of Cell Biology (Cambridge, MA: NPG Education)
[2]. Branden C I and Tooze J 2012 Introduction to Protein Structure, second edition (Boca Raton, FL: Garland Science)
[3]. Al-Lazikani B, Jung J, Xiang Z and Honig B 2001 Protein structure prediction Curr. Opin. Chem. Biol. 5 51–6
[4]. Kryshtafovych A, Schwede T, Topf M, Fidelis K and Moult J 2021 Critical assessment of methods of protein structure prediction (casp)—round xiv Proteins 89 1607–17
[5]. Jumper J et al. 2021 Highly accurate protein structure prediction with alphafold Nature 596 583–9
[6]. Gao M, Nakajima An D, Parks J M and Skolnick J 2022 Af2complex predicts direct physical interactions in multimeric proteins with deep learning Nat. Commun. 13 1744
[7]. Linger R M, Keating A K, Earp H S and Graham D K 2008 TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer Adv. Cancer Res. 100 35–83
[8]. Hynes N E and MacDonald G 2009 ErbB receptors and signaling pathways in cancer Curr. Opin. Cell Biol. 21 177–84
[9]. Graus-Porta D 1997 ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling EMBO J. 16 1647–55
[10]. Yarden Y 2001 Biology of her2 and its importance in breast cancer Oncology, suppl.S2 61 1–13
[11]. Wilding B et al. 2022 Discovery of potent and selective her2 inhibitors with efficacy against her2 exon 20 insertion-driven tumors, which preserve wild-type egfr signaling Nat. Cancer 3 pp. 821–36
[12]. Berman H M 2000 The protein data bank Nucleic Acids Res. 28 pp. 235–42
[13]. Bader G 2022 Her2 in complex with a covalent inhibitor wwPDB
[14]. Schrödinger, LLC 2015 The PyMOL molecular graphics system, version 1.8
[15]. Adasme M F, Linnemann K L, Bolz S N, Kaiser F, Salentin S, Haupt V, and Schroeder M 2021 Plip 2021: expanding the scope of the protein–ligand interaction profiler to dna and rna Nucleic Acids Res. 49 W530–4
Cite this article
Yao,W. (2024). Protein structure prediction based on deep learning: HER2 in complex with a covalent inhibitor. Advances in Engineering Innovation,6,13-20.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Journal:Advances in Engineering Innovation
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. O’Connor C M, Adams J U and Fairman J E 2010 Essentials of Cell Biology (Cambridge, MA: NPG Education)
[2]. Branden C I and Tooze J 2012 Introduction to Protein Structure, second edition (Boca Raton, FL: Garland Science)
[3]. Al-Lazikani B, Jung J, Xiang Z and Honig B 2001 Protein structure prediction Curr. Opin. Chem. Biol. 5 51–6
[4]. Kryshtafovych A, Schwede T, Topf M, Fidelis K and Moult J 2021 Critical assessment of methods of protein structure prediction (casp)—round xiv Proteins 89 1607–17
[5]. Jumper J et al. 2021 Highly accurate protein structure prediction with alphafold Nature 596 583–9
[6]. Gao M, Nakajima An D, Parks J M and Skolnick J 2022 Af2complex predicts direct physical interactions in multimeric proteins with deep learning Nat. Commun. 13 1744
[7]. Linger R M, Keating A K, Earp H S and Graham D K 2008 TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer Adv. Cancer Res. 100 35–83
[8]. Hynes N E and MacDonald G 2009 ErbB receptors and signaling pathways in cancer Curr. Opin. Cell Biol. 21 177–84
[9]. Graus-Porta D 1997 ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling EMBO J. 16 1647–55
[10]. Yarden Y 2001 Biology of her2 and its importance in breast cancer Oncology, suppl.S2 61 1–13
[11]. Wilding B et al. 2022 Discovery of potent and selective her2 inhibitors with efficacy against her2 exon 20 insertion-driven tumors, which preserve wild-type egfr signaling Nat. Cancer 3 pp. 821–36
[12]. Berman H M 2000 The protein data bank Nucleic Acids Res. 28 pp. 235–42
[13]. Bader G 2022 Her2 in complex with a covalent inhibitor wwPDB
[14]. Schrödinger, LLC 2015 The PyMOL molecular graphics system, version 1.8
[15]. Adasme M F, Linnemann K L, Bolz S N, Kaiser F, Salentin S, Haupt V, and Schroeder M 2021 Plip 2021: expanding the scope of the protein–ligand interaction profiler to dna and rna Nucleic Acids Res. 49 W530–4