







1. Introduction
Endocrine disruptors are substances that interfere with hormonal processes, such as hormone production, secretion, transport, receptor binding, or metabolism, thereby affecting physiological functions, including homeostasis, reproduction, development, and behavior [1]. BPA is extensively utilized in the inside coating of flatware, infant bottles, food storage containers, and food and beverage cans, and low-dose exposure to BPA has been demonstrated to interfere with many endocrine-related processes [2]. In adult mice, BPA impairs glucose homeostasis by rapidly lowering blood glucose and increasing insulin levels, eventually leading to chronic hyperinsulinemia and insulin resistance. Additionally, BPA interferes with synaptogenesis in the hippocampus and prefrontal cortex by blocking hormone-induced spine synapse formation, potentially impairing cognitive function [3]. As the BPA substitutes, there are also some concerns of BPS and BPF about endocrine disruption. Current studies on endocrine disruption of BPS and BPF are mostly about animals, such as zebrafish, mice, and so on instead focus on the human body[3, 4]. Molecular simulations and dynamics can simulate the binding of BPS, BPF, and human estrogen receptors, thereby inferring their interference with human endocrinology. This study utilize AutoDock Tools and AutoDock Vina to predict the binding sites of BPS and BPF in the estrogen receptor Ligand Binding Domain (LBD), use Gromacs 2021 to verify the stability of the interconnection result and calculate the binding free energy of BPA, BPS, and BPF to the receptor.
2. Materials and methods
2.1. Molecular docking: receptor protein/ligand small molecule structure acquisition and pretreatment
The receptor protein is Estrogen (PDB ID: 1A52) [5]. The protein structures were visualized and examined using PyMOL 3.0.3 software to ensure file integrity and docking suitability. The ligand files utilized in the study were acquired from the PubChem database and geometrically optimized by using the MMFF94 force field within OpenBabel software. The optimized molecular structure possesses the lowest energy and the ideal conformation, guaranteeing its suitability for subsequent molecular docking.
2.2. Molecular docking method
The receptor and ligand were produced utilizing AutoDock Tools 1.5.6. Polar hydrogen atoms were included into the receptor, and Gasteiger charges were allocated prior to storing the structure in pdbqt format. For the ligand, hydrogen atoms were also added and rotatable bonds were defined, and the optimized structure was saved in pdbqt format for docking. Grid parameters were set in the Grid module to ensure the active site was properly covered, with center coordinates defined as (X, Y, Z)=(108.1, 15.1, 96.0) and grid size set to (15.0×15.0×15.0). Following the empirical guideline that the grid size should be about 2.9 times the ligand Rg, a 15 Å box size was chosen to ensure sufficient coverage and flexibility for docking [6]. Molecular docking was conducted in semi-flexible mode with AutoDock Vina 1.2.0, with an exhaustiveness parameter set to 25 and the Lamarckian Genetic Algorithm chosen as the docking approach. The docking output included binding free energy values and docking pose files, which were used for further analysis and validation.
2.3. Molecule dynamics simulation methods
2.3.1. System construction and preparation
GROMACS 2021 was used to perform Molecular Dynamics (MD) simulations for 200 ns. The small-molecule ligands were subjected to the GAFF2 force field, while the protein was subjected to the Amber14SB force field. The SPC/E water model was used to solubilize each component in a periodic cubic water box with a 1.2 nm buffer. Na+ and Cl+ ions were added using a Monte Carlo ion insertion method in order to preserve charge neutrality. Utilizing the Particle Mesh Ewald (PME) technique, long-range electrostatic interactions were calculated.
2.3.2. Energy minimization and equilibration
Prior to production runs, the system underwent energy minimization and equilibration to guarantee structural stability. Energy minimization was conducted via the steepest descent algorithm for 50,000 iterations until the maximal force fell below 1000 kJ/mol. This was followed by two equilibration phases: an NVT ensemble at 310 K for 50,000 steps with a 2 fs timestep, and an NPT ensemble at 1 atm and 310 K which is for simulating Internal physiological environment for another 50,000 steps. These preparatory steps ensured thermal and pressure equilibrium before initiating the main simulation.
2.3.3. Production MD simulation and analysis
After minimization and equilibration, production molecular dynamics simulations were conducted for 200 ns with a 2 fs timestep. LINCS restrictions were implemented on all hydrogen-containing bonds. Trajectory coordinates were documented every 10 picoseconds for examination. Multiple structural and energetic parameters were calculated, including Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Radius of Gyration (Rg), Solvent-accessible Surface Area (SASA), and hydrogen bond count. Energy contributions from essential residues (within 4 Å of the ligand) and the total binding free energy were computed. Moreover, intricate structures were retrieved at five temporal intervals (0, 50, 100, 150, and 200 ns) to assess conformational alterations and binding stability during the simulation.
2.4. Calculation of binding free energy
The binding free energy of the receptor-ligand complexes was estimated using the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method. The final 10 ns of the equilibrated MD trajectories were analyzed using the gmx_MMPBSA tool to ensure representative sampling and reliable interaction evaluation. The binding free energy (
The total binding energy was decomposed into gas-phase energy (
Here,
In addition,
3. Results and discussion
3.1. Molecular docking results and analysis
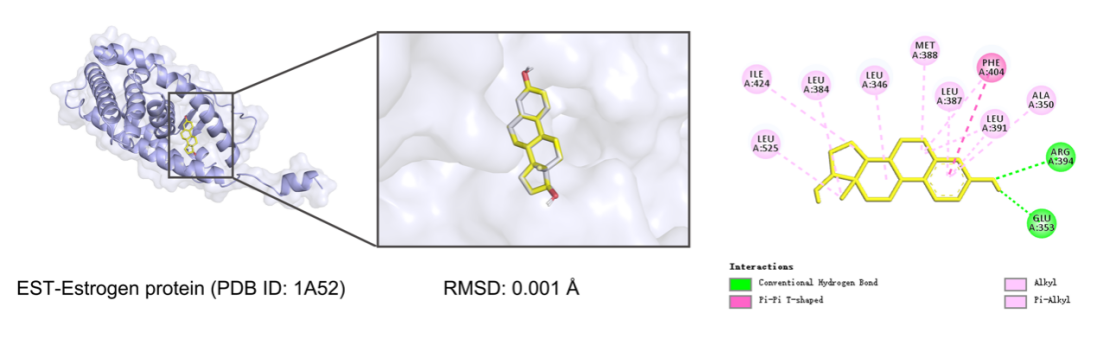
A redocking experiment was conducted utilizing the co-crystallized ligand EST to assess the reproducibility of the molecular docking methodology. The binding energy derived from the docking was −10.7 kcal/mol, signifying a robust and advantageous interaction between the ligand and the receptor. The Root Mean Square Deviation (RMSD) between the docked pose and the original crystallographic structure was computed to evaluate the precision of the docking procedure. As shown in figure 1, the resulting RMSD was 0.001 nm, which is significantly below the commonly accepted threshold of 2.0 nm. This exceptionally low RMSD value demonstrates that the docking method is capable of precisely predicting both the position and conformation of the ligand within the receptor binding site, the favorable binding energy and the high structural accuracy of the redocking results support the validity and reliability of the docking procedure used in this study.
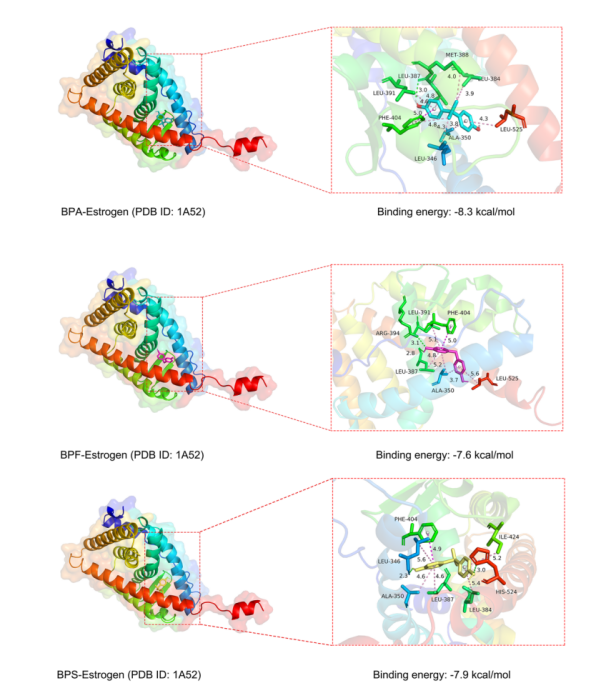
As shown in figure 2, in the BPA–estrogen receptor complex, BPA formed a hydrogen bond with the amino acid residue LEU-387 (0.3 nm), along with π-electron interactions with the receptor. In the BPF–estrogen receptor complex, BPF formed hydrogen bonds with ARG-394 (0.31 nm) and LEU-387 (0.28 nm), in addition to π-electron interactions. In the BPS–estrogen receptor complex, BPS formed hydrogen bonds with HIS-524 (0.3 nm) and LEU-346 (0.23 nm), while the sulfone group (O=S=O) of BPS also exhibited a π-sulfur interaction with the PHE-404 residue. Other non-covalent interactions primarily involved π-electron forces. These findings indicate that BPA, BPF, and BPS all bind to the estrogen receptor mainly through hydrogen bonding and π-electron interactions, with BPS showing an additional stabilizing π-sulfur interaction.
3.2. Molecular dynamics simulation results and analysis
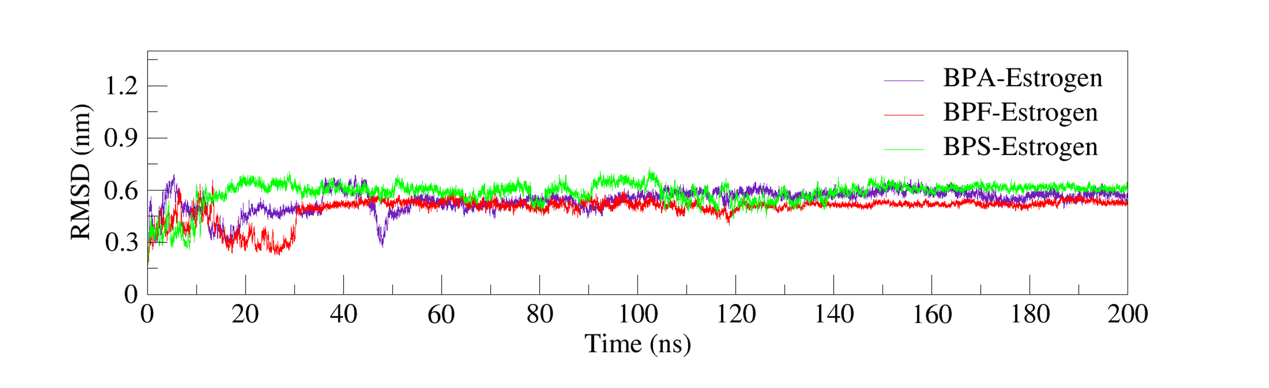
Root Mean Square Deviation (RMSD) is a commonly adopted metric for evaluating the structural stability of protein-ligand complexes during molecular dynamics simulations. Lower and stabilized RMSD values often imply minor structural variations and signal that the complex has reached a stable conformation [7].
In this study, molecular dynamics simulations were conducted to analyze the RMSD trajectories of BPA–Estrogen, BPF–Estrogen, and BPS–Estrogen complexes. As shown in figure 3, the RMSD of the BPA–estrogen complex gradually increased during the first 20 ns, reaching approximately 0.5 nm. Subsequently, it exhibited minor fluctuations over the next 30 ns and stabilized around 0.5 nm after 50 ns, and the RMSD of the BPA complex (0.5 nm) was significantly high compared to the crystal structure (EST-RMSD = 0.0001 nm), possibly reflecting local conformational adjustments upon ligand binding rather than overall structural destabilization. In contrast, the BPF–Estrogen and BPS–Estrogen complexes achieved relative stability around 30 ns, with final RMSD values maintained within the range of 0.5–0.6 nm. These results indicate that all three complexes attained structural stability during the simulations, demonstrating favorable binding stability.
Root Mean Square Fluctuation (RMSF) is an important parameter used to evaluate the flexibility and dynamic behavior of individual amino acid residues in proteins during Molecular Dynamics (MD) simulations. Higher RMSF values indicate greater conformational fluctuations, typically associated with flexible or disordered regions, whereas lower RMSF values suggest limited movement, often corresponding to rigid or structurally stable domains [8].
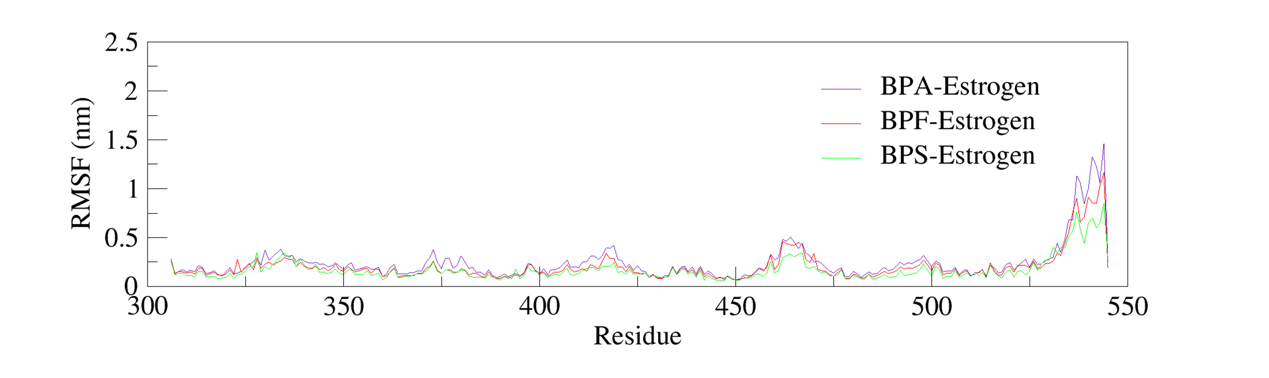
To assess local flexibility, RMSF profiles of the BPA–estrogen, BPF–estrogen, and BPS–estrogen complexes were analyzed. The results showed moderate fluctuations (below 0.5 nm) near residue positions 330, 375, 420, and 460, indicating moderate flexibility in these regions. As shown in figure 4, larger fluctuations (approximately 1.0–1.5 nm) were observed in the C-terminal α-helical region spanning residues 530–544, suggesting high conformational freedom in this terminal region. However, as this region does not constitute the active binding pocket, its mobility is unlikely to directly affect ligand binding stability.
Overall, in figure 5, the RMSF profiles of all three complexes exhibited similar trends. Residues located within the ligand-binding pocket showed consistently low RMSF values, indicating that ligand binding did not significantly disturb the rigid core of the protein. These findings further corroborate the structural stability of the receptor-ligand complexes throughout the simulation timeframe.
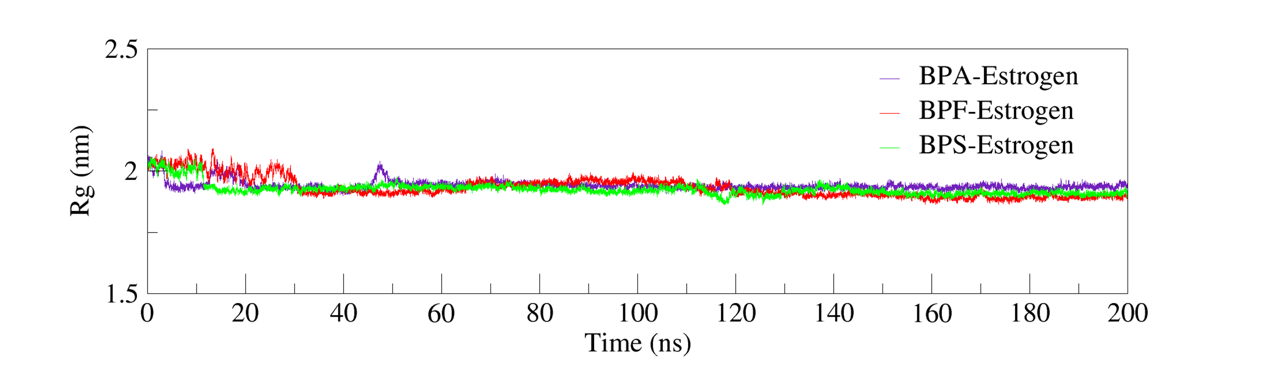
Radius of Gyration (Rg) is a key parameter used to assess the compactness and conformational stability of a protein or protein–ligand complex. Higher Rg values typically indicate structural expansion or relaxation during Molecular Dynamics (MD) simulations, while lower and stable Rg values reflect a compact conformation and greater structural integrity [9].
In figure 6, the Rg trajectories of all three complexes—BPA–Estrogen, BPF–Estrogen, and BPS–Estrogen—exhibited consistent stability over the simulation period, with no significant conformational expansion or collapse observed. The data indicate that ligand binding did not significantly alter the overall protein structure. The complexes retained a densely packed shape, signifying robust structural stability during the simulations.
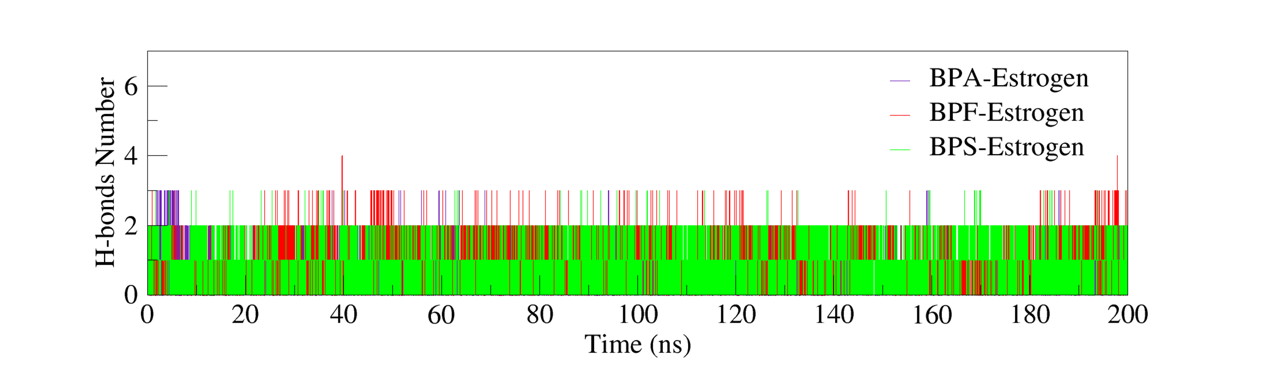
To investigate the hydrogen bonding characteristics at the binding sites of the complexes, the number of hydrogen bonds formed between the ligands and the receptor proteins during Molecular Dynamics (MD) simulations was calculated and analyzed. Hydrogen bonding is one of the primary non-covalent interactions that contributes to the structural stability of protein-ligand complexes. The number and temporal behavior of hydrogen bonds provide direct insight into the interaction strength and dynamic stability of the binding site.
Based on figure 7 of the 200 ns MD simulation data, both the BPA–Estrogen and BPF–Estrogen complexes stably maintained average two hydrogen bonds throughout the simulation. In contrast, the BPS–Estrogen complex formed one to two hydrogen bonds, with slight fluctuations in bond number over time. In comparison, the relatively fewer and less consistent hydrogen bonds in the BPS complex may contribute to slightly lower binding stability.
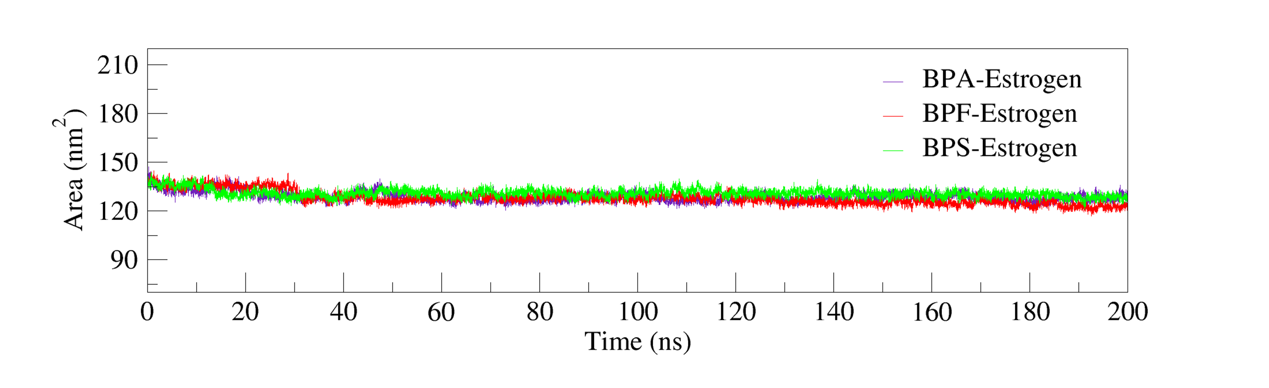
Solvent-Accessible Surface Area (SASA) is a critical parameter for evaluating protein conformational stability, folding state, and interactions with the solvent environment. Stability in SASA values is typically indicative of a structurally stable protein, whereas significant increases may suggest partial unfolding or conformational expansion. Conversely, a stable SASA trajectory implies that the protein retains its native fold without undergoing major structural rearrangement [10].
As shown in figure 8, the SASA analysis revealed that all three complexes—BPA–Estrogen, BPF–Estrogen, and BPS–Estrogen—exhibited stable SASA trajectories throughout the 200 ns molecular dynamics simulations. No significant fluctuations or long-term trends were observed. These findings indicate that ligand binding did not notably alter the overall solvent exposure of the receptor, and the complexes maintained stable conformational characteristics during the simulation period.
Upon achieving structural equilibrium in the complex systems, the Molecular Mechanics/generalized Born Surface Area (MM/GBSA) approach was utilized to calculate the binding free energies of various ligand-protein complexes. The MM/GBSA method disaggregates the binding free energy into molecular mechanics energy (including electrostatic and van der Waals interactions), solvation free energy (comprising polar and non-polar contributions), and, if included though typically disregarded, an entropy component. This method facilitates a quantitative evaluation of the binding stability between ligands and the receptor protein. The results in figure 9 indicated that the average binding free energies for the BPA–Estrogen, BPF–Estrogen, and BPS–Estrogen complexes were −33.00 kcal/mol, −34.45 kcal/mol, and −33.92 kcal/mol, respectively. All binding energies were negative, suggesting that each ligand spontaneously binds to the active site of the receptor, forming stable complexes. Typically, binding free energies in the range of −10 to −50 kcal/mol are considered indicative of favorable ligand–receptor interactions, further supporting the stability of the complexes observed in this study. Among them, the BPF–Estrogen complex exhibited the most favorable binding free energy (−34.45 kcal/mol), indicating its relatively higher binding affinity compared to BPA and BPS, and strong hydrophobic interactions of BPF (e.g., LEU346, LEU384) may be the key factor.
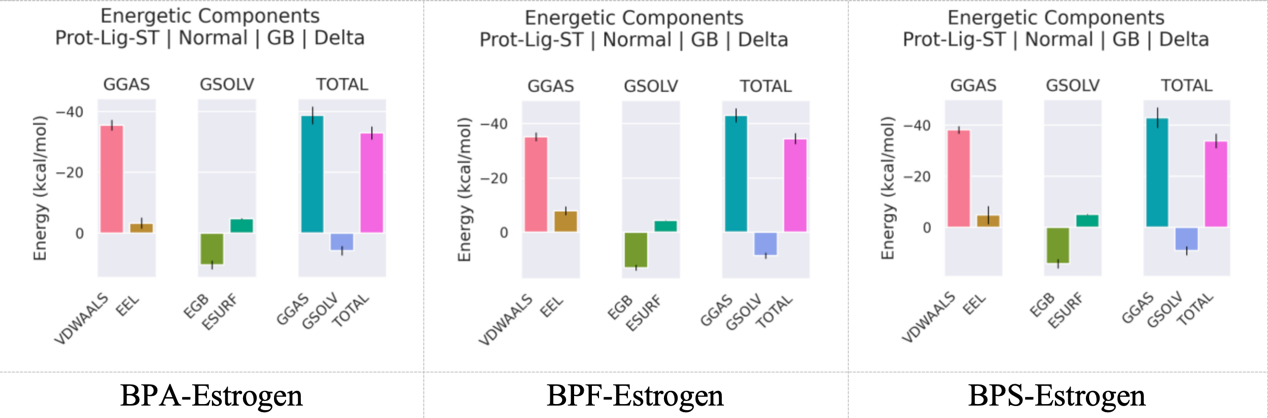
VDWAALS, EEL, EGB, ESURF, GGAS, GSOLV, and TOTAL refer respectively to van der Waals interactions, electrostatic interactions, polar solvation contributions, nonpolar solvation contributions, molecular mechanical energy, solvation energy, and the overall average binding free energy.
In conjunction with the analyses of hydrogen bonding, RMSD, RMSF, Radius of Gyration (Rg), and Solvent-Accessible Surface Area (SASA), the findings further illustrate that BPA, BPF, and BPS — functioning as estrogen analogs — exhibit significant binding affinity and structural stability at the binding site of the target protein.
These findings support the conclusion that all three compounds are capable of forming stable and energetically favorable complexes with the estrogen receptor throughout the molecular dynamics simulation timeframe.
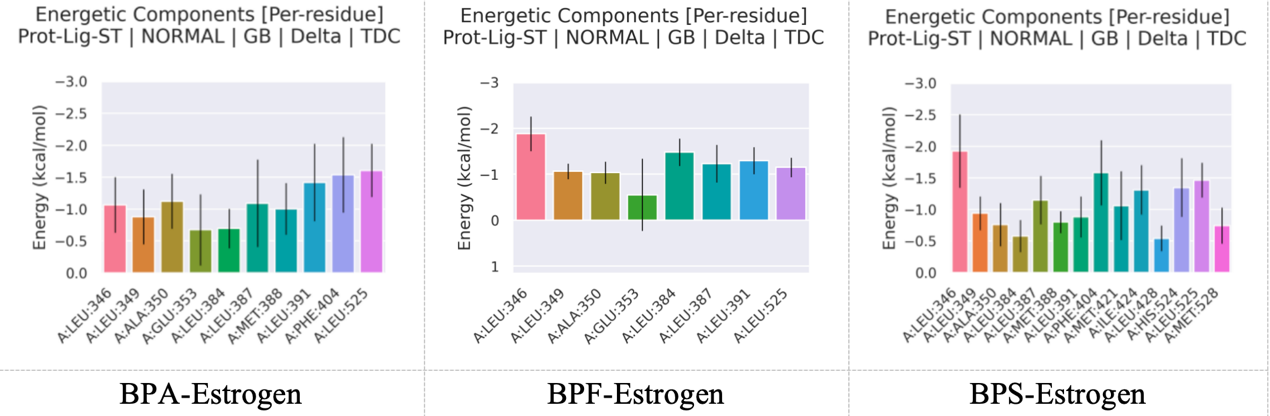
To further elucidate the interaction patterns of the BPA–Estrogen, BPF–Estrogen, and BPS–Estrogen complexes at the receptor binding site, residue energy decomposition analysis was performed based on the Molecular Dynamics (MD) trajectories. This analysis that shown in figure 10 provided insights into the interaction stability of each ligand at the binding site and identified the key amino acid residues contributing most significantly to the overall binding energy.
For the BPA–Estrogen complex, the major contributing residues were LEU346, LEU391, PHE404, and LEU525, with energy contributions of −1.07, −1.42, −1.54, and −1.61 kcal/mol, respectively. The strongest contributions from PHE404 and LEU525 suggest that hydrophobic interactions play a dominant role in stabilizing this complex.
In the BPF–Estrogen complex, key residues included LEU346 (−1.88 kcal/mol), LEU384 (−1.48 kcal/mol), and LEU525 (−1.15 kcal/mol). LEU346 showed the highest contribution, indicating strong hydrophobic interaction at this site. Compared to the BPA complex, the BPF complex exhibited slightly stronger overall residue contributions, especially at LEU346 and LEU384, suggesting a potentially higher binding affinity.
This may be attributed to sulfur–π interactions. Although less prevalent than other non-covalent interactions, such interactions have been identified in protein–ligand complexes and are recognized to contribute to binding stability.
The BPS–Estrogen complex involved a broader range of residues, including LEU346, PHE404, ILE424, and HIS524. The most significant contributions came from PHE404 (−1.58 kcal/mol) and HIS524 (−1.47 kcal/mol). Notably, MET421, ILE424, and HIS524 contributed markedly to the interaction, indicating a binding mode distinct from those of BPA and BPF, involving a wider array of amino acid residues.
4. Conclusion
Findings indicate that BPA, BPF, and BPS interact with the estrogen receptor's ligand-binding domain predominantly through hydrogen bonds and hydrophobic interactions, such as π-electron and π-sulfur interactions.All three complexes exhibited structural stability during 200 ns molecular dynamics simulations, with consistent radius of gyration (Rg) and solvent-accessible surface area (SASA) trajectories, indicating minimal conformational disruption. BPF displayed the strongest binding affinity, evidenced by its lowest binding free energy (-34.45 kcal/mol), driven by enhanced hydrophobic contributions from residues like LEU346 and LEU384. BPA and BPF complexes maintained more stable hydrogen bonding than BPS, which relied on diverse residue interactions (e.g., PHE404, HIS524) but showed slightly lower stability. Notably, all ligands remained anchored within the receptor’s active site throughout simulations, underscoring their persistent binding capability. These findings highlight that BPS and BPF, like BPA, exhibit robust binding to estrogen receptors, potentially posing similar endocrine-disruption risks. However, their distinct interaction profiles suggest varied mechanisms of action. Further investigations involving human studies are necessary to validate these computational findings. Future research should focus on elucidating the dose–response relationships of BPS and BPF, and on assessing their endocrine-disrupting effects in vivo. Moreover, the development and evaluation of safer BPA alternatives with minimized hormonal activity are warranted to address the potential health risks associated with current substitutes.
References
[1]. Kabir, E. R., Rahman, M. S., & Rahman, I. (2015). A review on endocrine disruptors and their possible impacts on human health.Environmental toxicology and pharmacology, 40(1), 241-258.
[2]. Huang, Y. Q., Wong, C. K. C., Zheng, J. S., Bouwman, H., Barra, R., Wahlström, B., ... & Wong, M. H. (2012). Bisphenol A (BPA) in China: a review of sources, environmental levels, and potential human health impacts.Environment international, 42, 91-99.
[3]. Qiu, W., Liu, S., Chen, H., Luo, S., Xiong, Y., Wang, X., ... & Wang, K. J. (2021). The comparative toxicities of BPA, BPB, BPS, BPF, and BPAF on the reproductive neuroendocrine system of zebrafish embryos and its mechanisms.Journal of hazardous materials, 406, 124303.
[4]. Meng, Z., Wang, D., Yan, S., Li, R., Yan, J., Teng, M., ... & Zhu, W. (2018). Effects of perinatal exposure to BPA and its alternatives (BPS, BPF and BPAF) on hepatic lipid and glucose homeostasis in female mice adolescent offspring.Chemosphere, 212, 297-306.
[5]. Li, L., Wang, Q., Zhang, Y., Niu, Y., Yao, X., & Liu, H. (2015). The molecular mechanism of bisphenol A (BPA) as an endocrine disruptor by interacting with nuclear receptors: insights from molecular dynamics (MD) simulations.PloS one, 10(3), e0120330.
[6]. Feinstein, W. P., & Brylinski, M. (2015). Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets.Journal of cheminformatics, 7, 1-10.
[7]. Aier, I., Varadwaj, P. K., & Raj, U. (2016). Structural insights into conformational stability of both wild-type and mutant EZH2 receptor.Scientific reports, 6(1), 34984.
[8]. Bagewadi, Z. K., Khan, T. Y., Gangadharappa, B., Kamalapurkar, A., Shamsudeen, S. M., & Yaraguppi, D. A. (2023). Molecular dynamics and simulation analysis against superoxide dismutase (SOD) target of Micrococcus luteus with secondary metabolites from Bacillus licheniformis recognized by genome mining approach.Saudi Journal of Biological Sciences, 30(9), 103753.
[9]. Lobanov, M. Y., Bogatyreva, N. S., & Galzitskaya, O. V. (2008). Radius of gyration as an indicator of protein structure compactness.Molecular Biology, 42, 623-628.
[10]. Ausaf Ali, S., Imtaiyaz Hassan, M., Islam, A., & Ahmad, F. (2014). A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states.Current Protein and Peptide Science, 15(5), 456-476.
Cite this article
Zhao,H. (2025). Molecular docking and dynamics reveal strong binding of BPA substitutes (BPS, BPF) to human estrogen receptor alpha: implications for endocrine disruption. Journal of Food Science, Nutrition and Health,4(1),62-69.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Journal:Journal of Food Science, Nutrition and Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Kabir, E. R., Rahman, M. S., & Rahman, I. (2015). A review on endocrine disruptors and their possible impacts on human health.Environmental toxicology and pharmacology, 40(1), 241-258.
[2]. Huang, Y. Q., Wong, C. K. C., Zheng, J. S., Bouwman, H., Barra, R., Wahlström, B., ... & Wong, M. H. (2012). Bisphenol A (BPA) in China: a review of sources, environmental levels, and potential human health impacts.Environment international, 42, 91-99.
[3]. Qiu, W., Liu, S., Chen, H., Luo, S., Xiong, Y., Wang, X., ... & Wang, K. J. (2021). The comparative toxicities of BPA, BPB, BPS, BPF, and BPAF on the reproductive neuroendocrine system of zebrafish embryos and its mechanisms.Journal of hazardous materials, 406, 124303.
[4]. Meng, Z., Wang, D., Yan, S., Li, R., Yan, J., Teng, M., ... & Zhu, W. (2018). Effects of perinatal exposure to BPA and its alternatives (BPS, BPF and BPAF) on hepatic lipid and glucose homeostasis in female mice adolescent offspring.Chemosphere, 212, 297-306.
[5]. Li, L., Wang, Q., Zhang, Y., Niu, Y., Yao, X., & Liu, H. (2015). The molecular mechanism of bisphenol A (BPA) as an endocrine disruptor by interacting with nuclear receptors: insights from molecular dynamics (MD) simulations.PloS one, 10(3), e0120330.
[6]. Feinstein, W. P., & Brylinski, M. (2015). Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets.Journal of cheminformatics, 7, 1-10.
[7]. Aier, I., Varadwaj, P. K., & Raj, U. (2016). Structural insights into conformational stability of both wild-type and mutant EZH2 receptor.Scientific reports, 6(1), 34984.
[8]. Bagewadi, Z. K., Khan, T. Y., Gangadharappa, B., Kamalapurkar, A., Shamsudeen, S. M., & Yaraguppi, D. A. (2023). Molecular dynamics and simulation analysis against superoxide dismutase (SOD) target of Micrococcus luteus with secondary metabolites from Bacillus licheniformis recognized by genome mining approach.Saudi Journal of Biological Sciences, 30(9), 103753.
[9]. Lobanov, M. Y., Bogatyreva, N. S., & Galzitskaya, O. V. (2008). Radius of gyration as an indicator of protein structure compactness.Molecular Biology, 42, 623-628.
[10]. Ausaf Ali, S., Imtaiyaz Hassan, M., Islam, A., & Ahmad, F. (2014). A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states.Current Protein and Peptide Science, 15(5), 456-476.