







1. Introduction
Chemistry today faces many experimental difficulties, including high costs, time-consuming, and difficult to duplicate. As the complexity of the experiments increases, the specialized chemical, reagents, and equipment required for the experiments become more expensive [1]. For example, the molecules and cells required in drug preparation experiments are very expensive. The molecules synthesized have varying degrees of purity, making it extremely difficult to maintain the same standards. In the case of the EGFR genome, culturing it as a research subject requires a long process, and its complexity makes it difficult to replicate the results of experiments.
Computational Chemistry involves using computational resources, physical theory for atomic-level chemistry, algorithms, and AI simulations to predict the behavior of chemical systems [2]. It complements experimental chemistry by providing insights into molecular structures, properties, and interactions at a molecular level [3]. With advances in software engineering, AI has improved its learning and information processing capabilities. With the aid of artificial intelligence, computational chemistry can encompass various techniques and methodologies that enable researchers to simulate chemical processes and properties without needing physical experiments [4].
Compared with traditional chemistry experiments, computational chemistry can save a lot of experimental resources, including experimental apparatus and the use of chemicals, making it a more cost-effective approach for exploring chemical phenomena [5]. Computer simulations allow researchers to gain a deeper understanding of chemical reactions, molecules’ behavior, and their interactions. The simulator will run through different environments and scenarios in a very short period. Computers will more intuitively show intricate molecular details and predict the possible molecular properties, behavior, and reactions through specific chemical models [6].
In the case of detection of EGFR mutation, computational chemistry can be used to study and predict the effects of mutations in the EGFR (epidermal growth factor receptor); the computer can help analyze the structural and functional impact of these mutations, predict how mutations in the EGFR gene alter the protein’s three-dimensional structure. It can be used as virtual screening and molecular docking to design drugs that target specific EGFR mutations6. The system can simulate multiple EGFR genomes and allow the comparison of different EGFR mutations to determine similarities in their effects on protein structure and function.
Computational Chemistry includes quantum chemistry simulation, molecular dynamic simulation, and AI generation of de novo molecules; the computer will process all the data input into the simulation and assist in analyzing complex experimental data [2]. Recent advances in machine learning and statistical inference have also shown potential in accelerating the discovery process in physical chemistry and related fields [1].
2. Principles of molecular dynamics simulations
2.1. Newton’s second law
Newton’s second law of motion, a fundamental principle in physics, provides a quantitative description of the influence of forces on the motion of objects. This law establishes a relationship between an object’s mass and the resulting acceleration. The equation F=ma, where F represents the force, m denotes mass in kilograms, and asignifies acceleration, embodies this principle.
2.2. Force Field
In the domain of molecular dynamics simulations, Newton’s second law of motion emerges as a fundamental cornerstone, playing a pivotal role in unraveling the intricate behaviors and interactions of molecules within a controlled environment. This law, which establishes a direct connection between the force applied to an object and its subsequent motion, proves essential for modeling the dynamic nature of atoms and molecules over time.
Central to this simulation methodology is the concept of a “force field.” Picture this as a conceptual scaffold—a virtual framework—constructed to encapsulate the multifaceted interplay of atomic and molecular dynamics within a predetermined system. This force field is designed to define the interactions between individual particles, such as atoms and molecules, through a series of mathematical equations and parameters. Those mathematical parameters are obtained from high accuracy quantum mechanics (QM) calculations, such as CCSDT, MP2, DFT, etc. These equations encapsulate the intricate intermolecular forces and energies governing the molecular system.
It’s noteworthy that the scientific community has crafted a variety of these force fields, each tailored to specific types of systems and molecular phenomena. These force fields have emerged as powerful tools to facilitate molecular dynamics simulations. By providing sets of precisely calibrated parameters, these force fields pave the way for researchers to define and explore the potential energy landscape governing the interactions among molecules.
For instance, prominent force fields like CHARMM, AMBER, and GROMOS have been established. These force fields are not arbitrary; rather, they’re the result of meticulous research, combining theoretical principles, empirical observations, and quantum mechanical insights. They provide a structured framework that guides simulations by governing how atoms and molecules move and interact in response to the forces acting upon them.
In essence, this concept of a force field acts as a computational microscope, allowing scientists to peer into the dynamic world of molecular interactions that operate at scales far beyond human observation. It serves as an indispensable tool for studying a wide range of molecular phenomena, including protein folding, drug binding, chemical reactions, and material properties [7]. Through the lens of Newton’s second law and guided by the principles encoded in force fields, molecular dynamics simulations empower scientists to uncover the nuanced choreography of molecular behavior, shedding light on the intricate workings of the molecular universe.
\( HΨ=EΨ\ \ \ (1) \)
\( \begin{array}{c} E=\sum _{bonds}{K_{b}}{(r-{r_{0}})^{2}} \\ +\sum _{angles}{K_{θ}}{(θ-{θ_{0}})^{2}}+\sum _{dihedral}{V_{n}}[1+cos{(nϕ-γ)}] \\ +\sum _{i=1}^{N-1}\sum _{j=i+1}^{N} \begin{array}{c} \ \ \ \\ \frac{{q_{i}}{q_{j}}}{{r_{ij}}}+{ϵ_{ij}} \end{array} [{(\frac{{R_{min,ij}}}{{r_{ij}}})^{2}}-2{(\frac{{R_{min,ij}}}{{r_{ij}}})^{6}}] \ \ \ (2) \end{array} \)
Equation (1) refers to the simplified format of the Time Independent Schrodinger Equation; equation (2) refers to the general form of force field; these equations refer to the force field of biological system[8].
One of the most prominent force fields is the CHARMM (Chemistry at HARvard Macromolecular Mechanics) force field. CHARMM is particularly well-suited for biomolecular simulations, such as proteins, nucleic acids, and lipids. Its parameters are tailored to accurately represent the complex behaviors and interactions observed in biological macromolecules. CHARMM excels in situations involving larger biomolecular systems and is often used for studying protein folding, molecular recognition, and enzyme catalysis due to its specialized treatment of non-bonded interactions and dihedral potentials [9]. As equation (3) shows, potential energy function of CHARMM22 is in the following form[10].
\( \begin{array}{c} V=\sum _{bonds}{k_{b}}{(b-{b_{o}})^{2}}+\sum _{angles}{k_{θ}}{(θ-{θ_{0}})^{2}} \\ +\sum _{dihedrals}{k_{ϕ[1+cos{(nϕ-δ)}]}}+\sum _{impropers}{k_{ω}}{(ω-{ω_{0}})^{2}} \\ +\sum _{Urey-Bradley}{k_{u}}{(u-{u_{0}})^{2}}+\sum _{nonbonded}({ϵ_{ij}}[{(\frac{{R_{min,ij }}}{{r_{ij}}})^{12}}-2{(\frac{{R_{min,ij}}}{{r_{ij}}})^{6}}]+\frac{{q_{i}}{q_{j}}}{{ϵ_{r}}{r_{ij}}})\ \ \ (3) \end{array} \)
AMBER (Assisted Model Building with Energy Refinement) force field is renowned for its versatility in simulating a broad range of biomolecular systems, from small organic molecules to large protein complexes. AMBER provides a balanced representation of bonded and non-bonded interactions, making it suitable for studying protein-ligand binding, protein-protein interactions, and nucleic acid dynamics. Its parameterization includes diverse systems, allowing researchers to explore various biophysical phenomena with confidence [11]. Equation (4) shows the functional form of AMBER force field [12].
\( \begin{array}{c} V({r^{N}})=\sum _{i∈bonds}{k_{bi}}{({l_{i}}-l_{i}^{0})^{2}}+\sum _{i∈angles}{k_{ai}}{({θ_{i}}-θ_{i}^{o})^{2}} \\ +\sum _{i∈torsions}\sum _{N}\frac{1}{2}V_{i}^{n}[1+cos{(n{ω_{i}}-{γ_{i}})}] \\ +\sum _{j=1}^{N-1}\sum _{i=j+1}^{N}{f_{ij}}\lbrace {∈_{ij}}[{(\frac{r_{ij}^{0}}{{r_{ij}}})^{12}}-2{(\frac{r_{ij}^{0}}{{r_{ij}}})^{6}}]+\frac{{q_{i}}{q_{j}}}{4π{ϵ_{0}}{r_{ij}}}\rbrace \ \ \ (4) \end{array} \)
On the other hand, the GROMOS (GROningen MOlecular Simulation) force field is often utilized for simulations involving smaller organic molecules and small peptides. GROMOS is notable for its accuracy in representing physical properties of simple molecules, making it suitable for studies on solvent effects, conformational analysis, and thermodynamics. It employs a different functional form for bonded interactions compared to other force fields, making it especially effective in situations where this distinction is crucial [13].
In summary, the choice of force field depends on the specific system under investigation. CHARMM is ideal for biomolecular simulations, AMBER offers versatility across a wide range of systems, and GROMOS is well-suited for smaller molecules. Researchers carefully select the appropriate force field based on the system’s characteristics and the phenomena they intend to explore. This tailored selection ensures that the molecular dynamics simulations provide meaningful insights into the behaviors and interactions within the chosen molecular system.
The principle of molecular dynamics represents a sophisticated interplay of numerical computation that unveils the dynamic orchestration of atoms and molecules. Governed by Newton’s second law of motion, this approach delves into the nanoscale, simulating the behavior of individual particles within a molecular framework. By treating atoms and molecules as dynamic entities, molecular dynamics elucidates the continuous interplay of forces that dictate their motion [14]. Force fields, acting as mathematical interpreters of molecular interactions, encode an extensive array of forces influencing molecular behavior, encompassing covalent bond dynamics, electrostatic interactions, and more. Through iterative calculations, particle positions and velocities are continuously adjusted, providing a vivid portrayal of the evolution of molecular structures over time [15]. This computational dance yields profound insights into intricate processes like protein folding, chemical reactions, and material characteristics, affording researchers the means to virtually explore the nuanced choreography underlying the molecular landscape.
3. Application of molecular dynamic simulation
Molecular dynamics (MD) simulation, with its ability to capture the temporal evolution of molecular systems, has become a cornerstone in computational chemistry. Its applications span a wide spectrum, from elucidating intricate biological processes to guiding the design of novel materials. In the realm of drug discovery and material design, specific force fields have been instrumental in providing insights that drive innovation.
3.1. Drug Discovery: Protein-Ligand Binding with the AMBER Force Field
HIV-1 protease, an enzyme integral to the HIV virus’s life cycle, has been a focal point in the quest for antiretroviral drugs. The enzyme’s role in the maturation of viral particles makes it a prime target for inhibition. Molecular dynamics simulations, especially those employing the AMBER force field, have been pivotal in demystifying the binding dynamics of potential inhibitors to the HIV-1 protease’s active site [16].
By harnessing the AMBER force field, the dynamic interplay between the protease and prospective drug molecules can be simulated. This facilitates the exploration of binding affinity, ligand stability, and specificity. Observations at the atomic scale reveal key residues pivotal for binding and potential allosteric sites that might modulate enzyme activity. Such insights, which encompass conformational shifts upon ligand binding and the role of water molecules in interactions, are invaluable in refining drug design, steering the synthesis of ligands for enhanced efficacy [17].
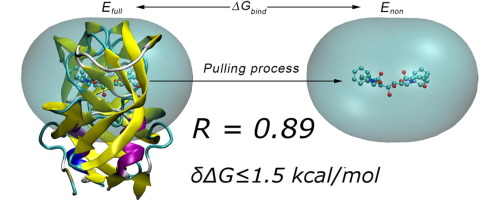
Figure 1. The calculation of ligand binding free energy for HIV-1 protease via AMBER Force Field [18].
3.2. Material Design: Crafting High-Performance Polymer Blends with the GROMOS Force Field
The versatility of polymers underpins their ubiquity in diverse applications, from electronics to packaging. The challenge lies in tailoring polymer blends to exhibit a desired set of mechanical, thermal, and optical properties. Molecular dynamics simulations, particularly those utilizing the GROMOS force field, offer a solution [19].
In the design of polymer blends, such as those combining a rigid, high-strength polymer with a flexible elastomer, the GROMOS force field can simulate the interplay between different polymer chains. These simulations shed light on molecular interactions, stress distribution, and the overall mechanical response of the material. By understanding how polymers intertwine and the nature of their interactions, the design process can be steered to achieve a blend that marries the rigidity of one component with the flexibility of another [20].
Simulating the blend under varied conditions, like different temperatures or strain rates, further refines the design process. It offers predictions on the polymer blend’s real-world behavior, guiding the choice of components and processing conditions to optimize performance [21].
In sum, the prowess of molecular dynamics simulations, anchored by force fields such as AMBER and GROMOS, has bridged theoretical constructs with experimental realities. Whether it’s the intricate world of drug discovery or the vast landscape of material design, these simulations have become an indispensable tool, driving innovation and expanding our understanding of molecular systems.
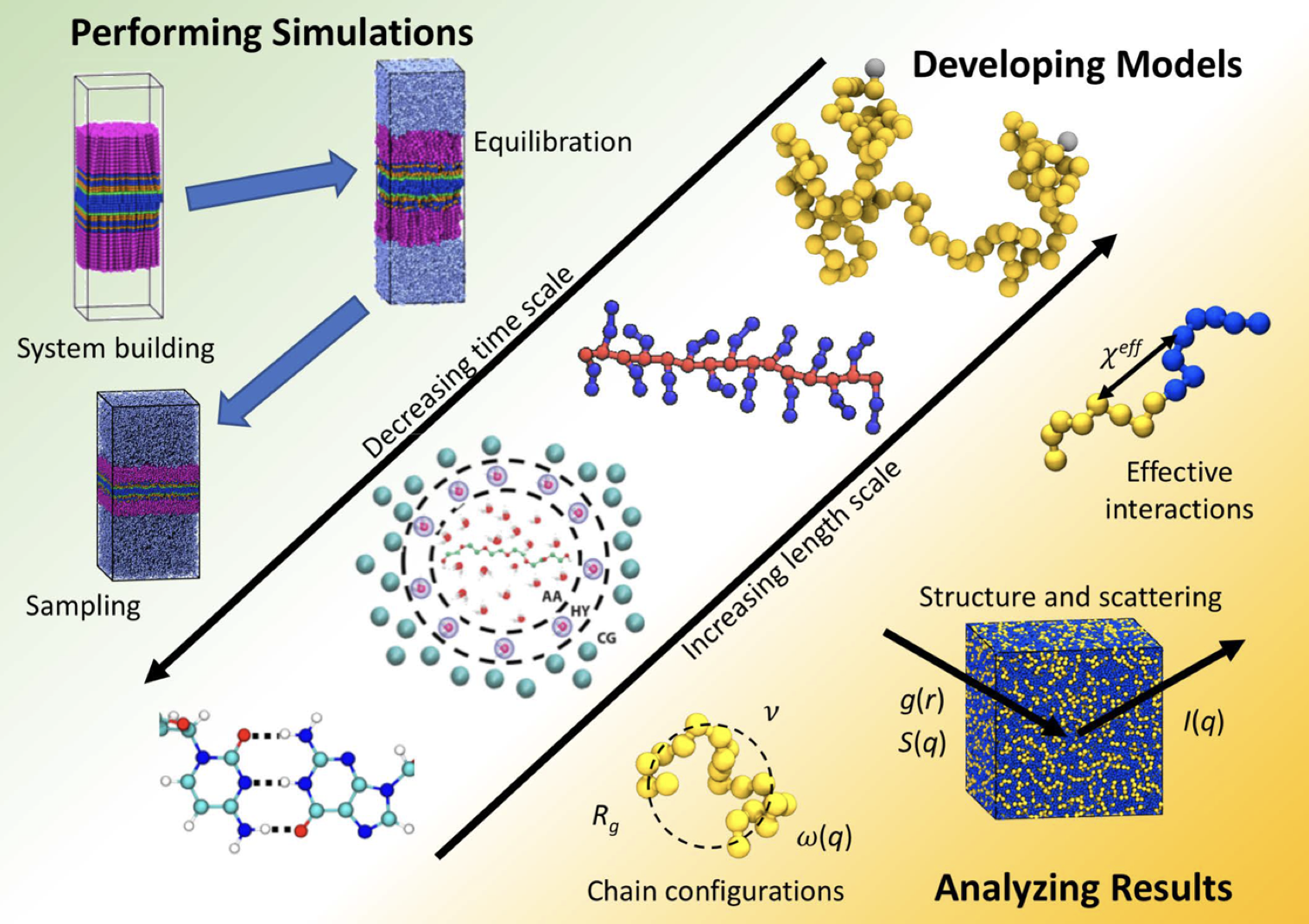
Figure 2. General guideline of material (polymer) development using MD simulation [22].
4. Artificial Intelligence for Chemistry
As the quest for understanding molecular phenomena has advanced, molecular dynamics (MD) simulations have been instrumental in offering detailed insights into complex chemical systems. However, despite the vast computational prowess these simulations command, the sheer complexity and scale of chemical data have also posed challenges. In recent times, a paradigm shift is being observed where traditional simulation approaches are converging with cutting-edge machine learning techniques. Artificial intelligence (AI) offers the potential to augment, and in some cases revolutionize, our understanding derived from MD simulations. By parsing massive datasets, optimizing simulation parameters, and even predicting molecular behavior, AI integrates with computational chemistry, promising a new era of enhanced understanding and predictive power.
4.1. AI for biology
AI has revolutionized drug discovery by significantly expediting the process of identifying potential drug candidates. For instance, Atomwise utilizes AI technology, AtomNet, to predict how different compounds will interact with biological targets, helping in the discovery of promising compounds for diseases like Ebola and multiple sclerosis. Traditional drug discovery involves synthesizing and testing countless molecules, a time-consuming and costly endeavor [23]. As Figure 2 shows, AI, powered by machine learning models, can analyze the chemical properties of millions of compounds and prioritize the most promising candidates for further experimental validation, reducing the risk of costly failures in the later stages of development.
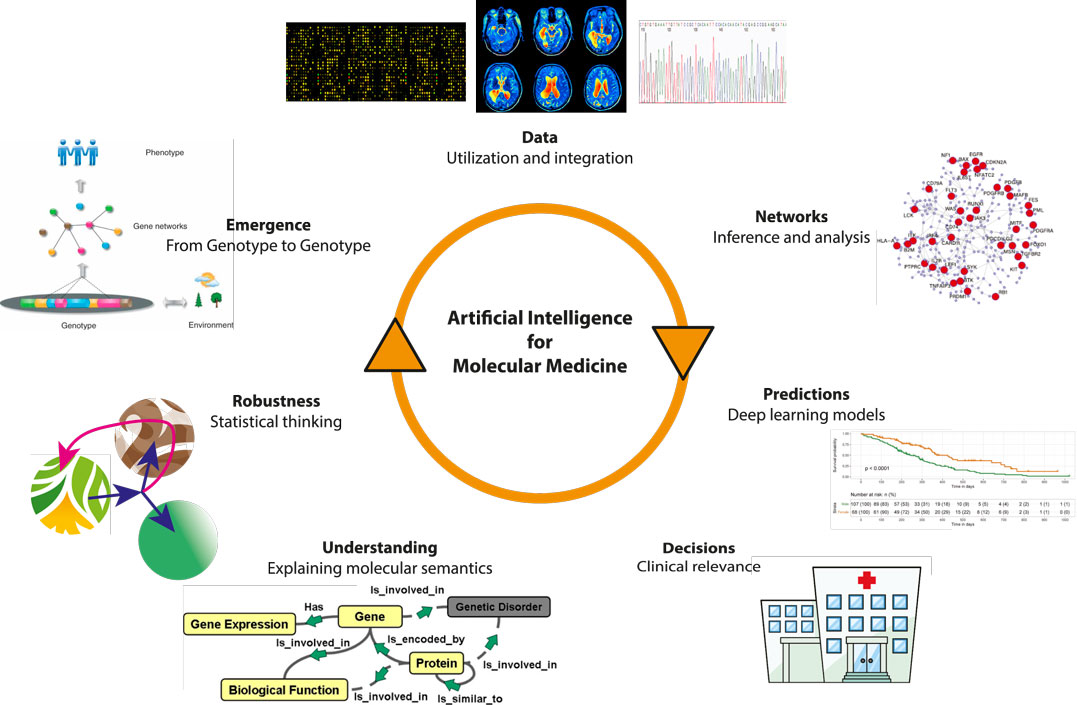
Figure 3. Artificail Intelligence for Molecular Medicine industry [24].
In the realm of biotechnology, AI-driven protein design is reshaping the field. DeepMind’s AlphaFold is a groundbreaking tool in this area, predicting the 3D structures of proteins with high accuracy, which is crucial for understanding diseases and developing new drugs [25]. Researchers can use computational tools like AlphaFold to modify and optimize protein structures for specific functions, such as designing enzymes with enhanced catalytic activity, applicable in biofuel production and pharmaceuticals. This not only accelerates drug discovery but also enables the creation of more effective therapeutics.
AI is transforming our understanding of diseases by analyzing complex biological data at an unprecedented scale. In genomics, Tempus is leveraging AI to analyze clinical and molecular data, providing insights that help doctors make more personalized treatment plans for cancer patients. AI algorithms identify genetic mutations associated with diseases like cancer and pinpoint potential targets for therapy. In personalized medicine, AI analyzes an individual’s genetic makeup and medical history to tailor treatment plans, optimizing therapeutic outcomes. Additionally, AI-driven image analysis enhances medical imaging techniques, such as MRI and CT scans, improving disease diagnosis and monitoring.
Handling vast datasets has always been a challenge in the life sciences, given the sheer volume of biological information generated daily. AI excels at managing and extracting valuable insights from these datasets. Machine learning algorithms identify biomarkers associated with diseases, track disease progression, and predict patient outcomes. In epidemiology, AI analyzes data on disease prevalence, demographics, and environmental factors to predict outbreaks and inform public health strategies [26]. This data-driven approach is invaluable in understanding the complex dynamics of diseases and guiding evidence-based decision-making.
As AI continues to advance in biology, it not only enhances the efficiency and precision of research but also opens up entirely new avenues for scientific exploration. The synergy between AI and biology holds the potential to accelerate discoveries, drive innovation in healthcare, and ultimately improve the well-being of individuals and populations worldwide.
AI’s transformative influence extends seamlessly into material science, reshaping the landscape of material discovery. Traditionally, the search for novel materials involved laborious trial-and-error experimentation. However, AI-driven approaches, honed in the biological sciences, are fundamentally altering this process. Machine learning algorithms can now be adapted to analyze the properties of existing materials, predict variations in composition and structure, and pinpoint materials with desired characteristics, such as Citrine Informatics, which utilizes AI to accelerate the development of new materials. This breakthrough accelerates the identification of materials with specific attributes, such as superconductivity, high strength, or exceptional thermal conductivity, revolutionizing our ability to innovate in fields ranging from energy storage to aerospace.
4.2. AI for material science
AI’s profound influence in biology finds seamless integration with material science, particularly in the domain of material discovery. Building on its success in biology, AI has redefined the process of discovering novel materials. By analyzing extensive datasets of known materials and recognizing patterns within their properties, compositions, and structures, AI accelerates the identification of promising candidates for synthesis [27]. This transformative approach enhances efficiency across various fields, from electronics to renewable energy, by predicting material properties and guiding the development of innovative technologies.
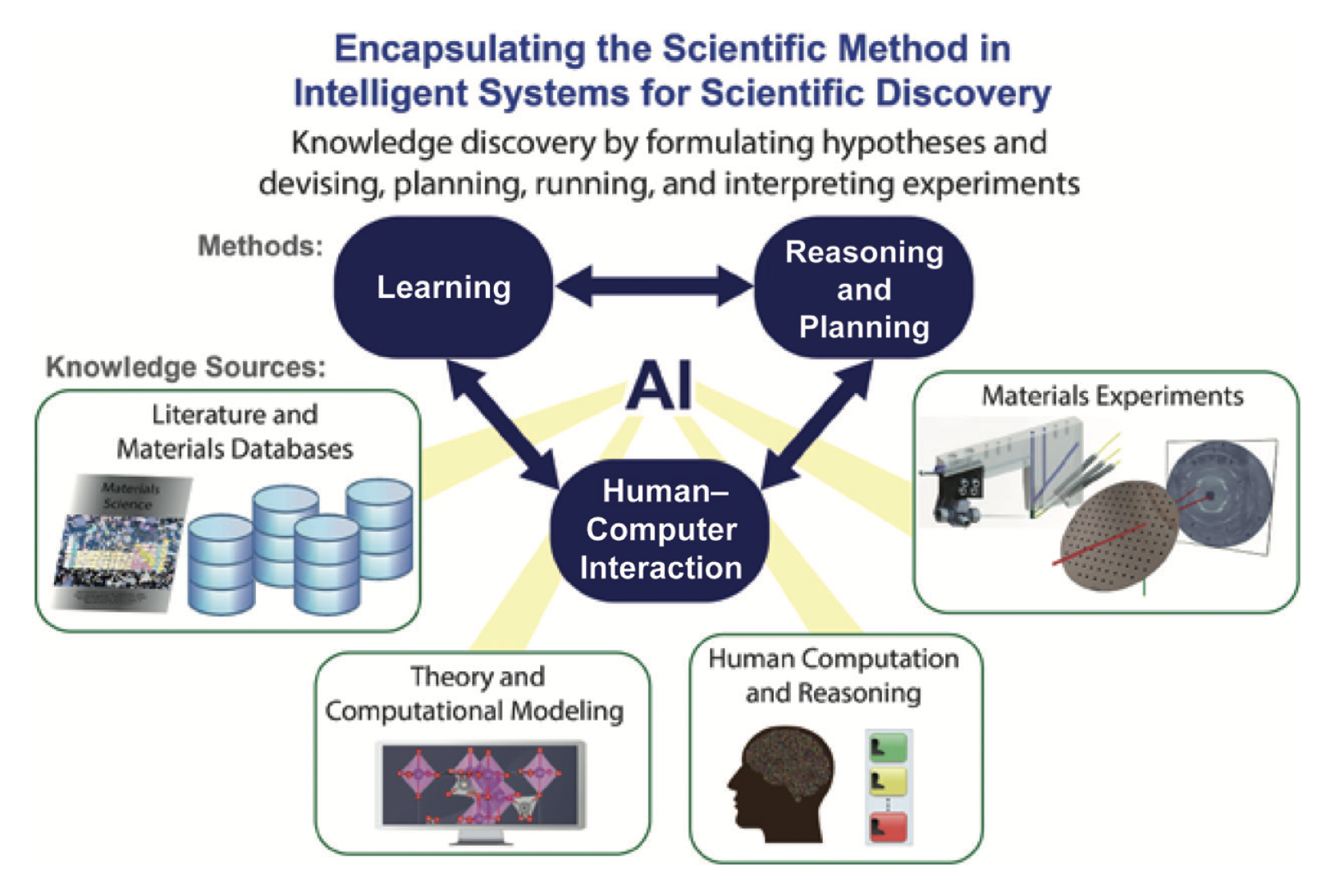
Figure 4. Artificail Intelligence for Material discovery [28].
Figure 4 depicts a conceptual framework for integrating Artificial Intelligence (AI) into the scientific method, specifically in the context of intelligent systems for scientific discovery of material within computational chemistry. At the core of this framework is the interplay between AI and human-computer interaction, facilitating enhanced learning, reasoning, and planning for knowledge discovery. AI draws on various knowledge sources, such as literature and materials databases, as well as theoretical and computational models, to formulate hypotheses and design experiments. The bidirectional arrows suggest a dynamic, iterative process where learning informs reasoning and vice versa, signifying that as the AI system acquires new data from materials experiments and human computation and reasoning, it refines its learning algorithms and planning strategies. This represents an advanced approach to scientific inquiry where computational methods augment traditional experiments, allowing for more efficient and accurate predictions and analyses in materials science and chemistry.
AI’s impact extends beyond discovery and into materials design, where researchers leverage AI to craft materials with precisely tailored properties. By specifying desired characteristics, AI algorithms generate molecular structures that meet these criteria, leading to materials customized for specific industrial applications, environmental considerations, and healthcare needs [29].
The integration of Artificial Intelligence (AI) in material design is heralding unprecedented advancements, particularly in the synthesis of materials characterized by enhanced sustainability, durability, and biocompatibility. This innovative approach is catalyzing transformative developments across diverse sectors, including but not limited to automotive manufacturing and healthcare.
The AI models employed in this research are capable of predicting the properties of materials prior to their synthesis, thereby significantly accelerating the material discovery process [30]. This acceleration is pivotal, enabling rapid innovations and advancements across various industrial domains. The predictive capabilities of AI not only streamline the material design process but also open avenues for the exploration of materials with unprecedented properties, thereby expanding the horizons of material science. The implications of such advancements are profound, promising a future where the synergy between AI and material science can drive the evolution of materials with tailored properties, catering to the nuanced needs of diverse applications, and contributing to the realization of a sustainable and technologically advanced future.
The collaboration between AI and material science not only accelerates innovation but also empowers researchers to explore uncharted territories. AI-driven simulations predict material behavior under extreme conditions, making advancements in aerospace and space exploration possible. Additionally, AI optimizes material compositions to enhance performance and longevity, offering benefits across diverse sectors, including construction, manufacturing, and energy storage. This partnership between AI and material science is shaping the future of materials and technology, promising to redefine possibilities and drive progress in various fields [31].
4.3. AI for Molecular Dynamic Simulation: Balance between accuracy and efficiency
In the realm of molecular dynamics (MD) simulations, achieving a delicate equilibrium between accuracy and efficiency is paramount. While classical MD simulations have proven invaluable in elucidating molecular behaviors, they often demand substantial computational resources and time. As a solution to this dilemma, researchers are increasingly turning to artificial intelligence (AI) to enhance the speed and efficiency of MD calculations while maintaining accuracy.
One promising avenue in this endeavor is the use of Deep Potential Molecular Dynamics (DeepMD) kits. These toolkits harness the power of deep learning and neural networks to revolutionize the way force fields are constructed and utilized in MD simulations. Unlike traditional force fields, which rely on handcrafted mathematical expressions and parameters, DeepMD kits leverage AI to learn and predict molecular interactions directly from data. For instance, in a theoretical investigation of the water phase diagram, a challenging endeavor due to the requirement for a highly accurate model of water interatomic interactions, researchers utilized the DeepMD-kit to construct a deep potential model for the water system across a diverse range of thermodynamic states from 0 to 2400 K and 0–50 GPa. The model, trained on density functional theory (DFT) data using the SCAN approximation of the exchange–correlation functional, exhibited a consistent accuracy within the relevant thermodynamic range, with an root mean square error (RMSE) of less than 1 meV/H2O. It accurately predicted fluid, molecular, and ionic phases, along with almost all stable ice polymorphs within the range, barring phases III and XV. This application not only validated the software implementation necessary for molecular dynamics simulations used in phase diagram calculations, but also demonstrated the potent capability of DeepMD kits in predicting water molecule ionization and understanding the atomistic mechanism of proton diffusion, thus exemplifying the transformative impact of AI on molecular dynamic simulations [32].
DeepMD kits work by training neural networks on large datasets of molecular configurations and their corresponding energies and forces. These trained models can then provide accurate predictions of energy and forces for new molecular configurations, effectively bypassing the need for time-consuming quantum mechanical calculations [33].
The advantage of DeepMD kits lies in their ability to capture complex, non-linear interactions between atoms and molecules. They excel in systems where traditional force fields may fall short in accuracy. Furthermore, these AI-based force fields are transferable, meaning they can often be applied to various systems, making them versatile tools for a wide range of MD simulations.
While AI-based force fields hold immense promise in accelerating MD simulations, it’s important to recognize that their use in scientific research, such as biology and materials, can introduce a potential trade-off between accuracy and efficiency. AI models, while powerful, are not infallible and may occasionally yield inaccurate predictions of biological or material properties.
In biological research, where AI aids in drug discovery, protein design, and disease understanding, the balance between accuracy and efficiency is crucial. AI models can rapidly screen vast chemical libraries to identify potential drug candidates, significantly speeding up the drug development process. However, there is always the risk that an AI model may overlook certain interactions or make predictions based on biased training data [34].
Similarly, in materials science, AI-driven material discovery and design have transformed the field. AI can predict novel materials with specific properties, reducing the need for extensive experimentation. However, there’s a need for rigorous validation and experimentation to ensure that the predicted materials meet real-world expectations.
The amalgamation of Artificial Intelligence (AI) within Molecular Dynamics (MD) simulations represents a seminal advancement, addressing the inherent challenges related to computational efficiency and time constraints prevalent in traditional simulation methodologies. This integration is pivotal, serving to significantly attenuate the computational and temporal requisites traditionally associated with MD simulations, thereby acting as a catalyst in the exploration of more extensive and intricate systems which were previously unattainable due to the limitations intrinsic to classical MD methods.
AI models, when meticulously trained, serve as sophisticated approximators of potential energy surfaces and force fields, thereby acting as surrogate models capable of representing intricate physical interactions with remarkable precision and accuracy. These models facilitate a deeper, more nuanced understanding of molecular behaviors and interactions, achieved at an accelerated pace, by rapidly learning from the data generated by MD simulations and refining their predictions and approximations iteratively.
This acceleration is of paramount importance, especially in the context of large-scale systems such as biomolecular complexes or materials characterized by a multitude of atoms, where conventional MD simulations are often impeded by extensive computational loads due to the complexity and scale of the systems under consideration. AI-driven models mitigate this impediment by efficiently navigating the high-dimensional space of molecular configurations and rapidly pinpointing regions of interest, thus enabling more focused and efficient simulations.
Furthermore, the integration of AI within MD simulations enables the exploration of a broader parameter space and facilitates the study of events occurring over longer timescales, which are typically computationally prohibitive with classical MD methods. This expanded capability is crucial for the elucidation of slow biological processes and the identification of rare events, which are integral for a comprehensive understanding of molecular mechanisms and are instrumental in the design of novel materials and pharmaceutical compounds.
By training AI models to approximate potential energy surfaces and force fields, scientists can gain a deeper understanding of molecular behavior and interactions at a faster pace. This acceleration is particularly beneficial in large-scale systems, such as biomolecular complexes or materials with numerous atoms.
5. Conclusion
In conclusion, the symbiotic relationship between Computational Chemistry and Artificial Intelligence stands as a testament to the incredible advancements within the realm of molecular research. This integration is not merely a technological upgrade but a paradigm shift that offers profound insights into molecular behavior and interactions. The utilization of AI algorithms in conjunction with molecular dynamics simulations has proven to be a formidable force, driving forward the efficiency and precision of research endeavors. The tangible benefits of this fusion are manifold, ranging from the accelerated discovery of pharmaceuticals to the creation of innovative materials that could revolutionize industries.
Despite the promise this union holds, it is not without its limitations. One of the primary constraints is the need for extensive computational resources that can handle complex algorithms and vast datasets inherent in AI-driven research. Additionally, there remains a learning curve for researchers integrating AI into their workflows, necessitating a cross-disciplinary approach that melds computational expertise with scientific inquiry.
Looking towards the future, it is anticipated that advancements in AI will continue to augment the capabilities of computational chemistry, with machine learning models becoming ever more sophisticated and capable of handling the nuanced subtleties of molecular science. The development of more intuitive AI systems could also democratize high-level molecular research, making it more accessible to a broader scientific community. As we move forward, it is essential that the research community remains vigilant, ensuring that ethical considerations keep pace with technological advancements.
The horizon of molecular research is expanding, and with it, our ability to confront and overcome the challenges of the future. As we refine these computational tools and broaden our understanding, we are not just witnessing the evolution of research methodologies but are actively participating in a scientific renaissance that promises to reshape our world.
References
[1]. Jadrich, R. B., Lindquist, B. A., & Truskett, T. M. (2017). Recent advances in accelerated discovery through machine learning and statistical inference
[2]. McArdle, S.; Endo, S.; Aspuru-Guzik, A.; Benjamin, S. C.; Yuan, X. Quantum Computational Chemistry. Rev. Mod. Phys. 2020, 92 (1), 015003. DOI: 10.1103/RevModPhys.92.015003.
[3]. Shikano, Y., Watanabe, H.C., Nakanishi, K.M. et al. Post-Hartree–Fock method in quantum chemistry for quantum computer. Eur. Phys. J. Spec. Top. 230, 1037–1051 (2021). https://doi.org/10.1140/epjs/s11734-021-00087-z
[4]. John A. Keith, Valentin Vassilev-Galindo, Bingqing Cheng, Stefan Chmiela, Michael Gastegger, Klaus-Robert Müller, and Alexandre Tkatchenko Chemical Reviews 2021 121 (16), 9816-9872 DOI: 10.1021/acs.chemrev.1c00107
[5]. Hanwell, M.D., de Jong, W.A. & Harris, C.J. Open chemistry: RESTful web APIs, JSON, NWChem and the modern web application. J Cheminform 9, 55 (2017). https://doi.org/10.1186/s13321-017-0241-z
[6]. G. Li, Y. Shi and A. Javadi-Abhari, “Software-Hardware Co-Optimization for Computational Chemistry on Superconducting Quantum Processors,” 2021 ACM/IEEE 48th Annual International Symposium on Computer Architecture (ISCA), Valencia, Spain, 2021, pp. 832-845, doi: 10.1109/ISCA52012.2021.00070
[7]. Cornell, W., Cieplak, P., Bayly, C., Gould, I., Merz, K., Ferguson, D., Spellmeyer, D., Fox, T., Caldwell, J. & Kollman, P. (1995). A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. Journal of the American Chemical Society, 117, 5179—5197
[8]. Liebl, K., & Zacharias, M. (2021). Tumuc1: A New Accurate DNA Force Field Consistent with High-Level Quantum Chemistry. Journal of Chemical Theory and Computation, 17(11), 7096-7105. https://doi.org/10.1021/acs.jctc.1c00682
[9]. Brooks, B. R., Brooks, C. L., 3rd, Mackerell, A. D., Jr, Nilsson, L., Petrella, R. J., Roux, B., Won, Y., Archontis, G., Bartels, C., Boresch, S., Caflisch, A., Caves, L., Cui, Q., Dinner, A. R., Feig, M., Fischer, S., Gao, J., Hodoscek, M., Im, W., Kuczera, K., … Karplus, M. (2009). CHARMM: the biomolecular simulation program. Journal of computational chemistry, 30(10), 1545–1614. https://doi.org/10.1002/jcc.21287
[10]. MacKerell Jr, A. D., Bashford, D., Bellott, M. L. D. R., Dunbrack Jr, R. L., Evanseck, J. D., Field, M. J., ... & Karplus, M. (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. The journal of physical chemistry B, 102(18), 3586-3616.
[11]. Case, D. A., Cheatham, T. E., 3rd, Darden, T., Gohlke, H., Luo, R., Merz, K. M., Jr, Onufriev, A., Simmerling, C., Wang, B., & Woods, R. J. (2005). The Amber biomolecular simulation programs. Journal of computational chemistry, 26(16), 1668–1688. https://doi.org/10.1002/jcc.20290
[12]. Cornell, W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, K. M., Ferguson, D. M., ... & Kollman, P. A. (1995). A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. Journal of the American Chemical Society, 117(19), 5179-5197
[13]. Nester, K., Gaweda, K., & Plazinski, W. (2019). A GROMOS Force Field for Furanose-Based Carbohydrates. Journal of chemical theory and computation, 15(2), 1168–1186. https://doi.org/10.1021/acs.jctc.8b00838
[14]. Ditler, E, Luber, S. Vibrational spectroscopy by means of first-principles molecular dynamics simulations. WIREs Comput Mol Sci. 2022; 12:e1605. https://doi.org/10.1002/wcms.1605
[15]. Ponder, J. W., & Case, D. A. (2003). Force fields for protein simulations. Advances in protein chemistry, 66, 27-85
[16]. He, X., Liu, S., Lee, T. S., Ji, B., Man, V. H., York, D. M., & Wang, J. (2020). Fast, accurate, and reliable protocols for routine calculations of protein–ligand binding affinities in drug design projects using AMBER GPU-TI with ff14SB/GAFF. ACS omega, 5(9), 4611-4619
[17]. Dong, X., Yuan, X., Song, Z., & Wang, Q. (2021). The development of an Amber-compatible organosilane force field for drug-like small molecules. Physical Chemistry Chemical Physics, 23(22), 12582-12591
[18]. Ngo, S. T., Nguyen, M. T., & Nguyen, M. T. (2017). Determination of the absolute binding free energies of HIV-1 protease inhibitors using non-equilibrium molecular dynamics simulations. Chemical Physics Letters, 676, 12-17. DOI: 10.1016/j.cplett.2017.03.034
[19]. Lins, R. D., & Hünenberger, P. H. (2005). A new GROMOS force field for hexopyranose‐based carbohydrates. Journal of computational chemistry, 26(13), 1400-1412
[20]. de Arenaza, I. M., Meaurio, E., & Sarasua, J. R. (2012). Analysis of the miscibility of polymer blends through molecular dynamics simulations. Polymerization
[21]. He, M., Qiu, F., & Lin, Z. (2013). Towards high-performance polymer-based thermoelectric materials. Energy & Environmental Science, 6(5), 1352-1361
[22]. Gartner III, T. E., & Jayaraman, A. (2019). Modeling and Simulations of Polymers: A Roadmap. Macromolecules, 52(3), 755-786. DOI: 10.1021/acs.macromol.8b01836
[23]. Maryasin, B., Marquetand, P., & Maulide, N. (2018). Machine learning for organic synthesis: are robots replacing chemists?. Angewandte Chemie International Edition, 57(24), 6978-6980
[24]. Emmert-Streib, F. (2021). Grand Challenges for Artificial Intelligence in Molecular Medicine. Frontiers in Molecular Medicine, 1. DOI: 10.3389/fmmed.2021.734659
[25]. Kim, H., Kim, E., Lee, I., Bae, B., Park, M., & Nam, H. (2020). Artificial intelligence in drug discovery: a comprehensive review of data-driven and machine learning approaches. Biotechnology and Bioprocess Engineering, 25, 895-930
[26]. Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O., & Walsh, A. (2018). Machine learning for molecular and materials science. Nature, 559(7715), 547-555
[27]. Zubarev, D., Mendes, C. R., Brazil, E. V., Cerqueira, R., Schmidt, K., Segura, V., ... & Sanders, D. (2022). Toward Human-AI Co-creation to Accelerate Material Discovery. arXiv preprint arXiv:2211.04257
[28]. Gomes, C.P., Selman, B. & Gregoire, J.M. Artificial intelligence for materials discovery.MRS Bulletin 44, 538–544 (2019). DOI: 10.1557/mrs.2019.158
[29]. Kailkhura, B., Gallagher, B., Kim, S., Hiszpanski, A., & Han, T. Y. J. (2019). Reliable and explainable machine-learning methods for accelerated material discovery. npj Computational Materials, 5(1), 108
[30]. Qayyum, F., Kim, D. H., Bong, S. J., Chi, S. Y., & Choi, Y. H. (2022). A Survey of Datasets, Preprocessing, Modeling Mechanisms, and Simulation Tools Based on AI for Material Analysis and Discovery. Materials, 15(4), 1428
[31]. Zylla, J. L., Gnimpieba, E., Bomgni, A. B., Sani, R. K., Subramaniam, M., Lushbough, C., ... & Chundi, P. (2023). Convergence research and training in computational bioengineering: a case study on AI/ML driven biofilm-material interaction discovery
[32]. Zhang, L., Han, J., Wang, H., Car, R., & Weinan, E. J. P. R. L. (2018). Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Physical review letters, 120(14), 143001
[33]. Zhang, L., Huang, K., Yang, Y. I., & Shi, L. (2022). Combined artificial intelligence and molecular dynamics (AI-MD) methods. Frontiers in molecular biosciences, 9, 1012785
[34]. Ye, F., Liang, Z., & Broussy, Y. (Eds.). (2022). Computer-aided drug design: Drug discovery, computational modelling, and artificial intelligence
Cite this article
Zhuo,Y. (2024). Computational chemistry review article. Applied and Computational Engineering,61,262-273.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Materials Chemistry and Environmental Engineering
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Jadrich, R. B., Lindquist, B. A., & Truskett, T. M. (2017). Recent advances in accelerated discovery through machine learning and statistical inference
[2]. McArdle, S.; Endo, S.; Aspuru-Guzik, A.; Benjamin, S. C.; Yuan, X. Quantum Computational Chemistry. Rev. Mod. Phys. 2020, 92 (1), 015003. DOI: 10.1103/RevModPhys.92.015003.
[3]. Shikano, Y., Watanabe, H.C., Nakanishi, K.M. et al. Post-Hartree–Fock method in quantum chemistry for quantum computer. Eur. Phys. J. Spec. Top. 230, 1037–1051 (2021). https://doi.org/10.1140/epjs/s11734-021-00087-z
[4]. John A. Keith, Valentin Vassilev-Galindo, Bingqing Cheng, Stefan Chmiela, Michael Gastegger, Klaus-Robert Müller, and Alexandre Tkatchenko Chemical Reviews 2021 121 (16), 9816-9872 DOI: 10.1021/acs.chemrev.1c00107
[5]. Hanwell, M.D., de Jong, W.A. & Harris, C.J. Open chemistry: RESTful web APIs, JSON, NWChem and the modern web application. J Cheminform 9, 55 (2017). https://doi.org/10.1186/s13321-017-0241-z
[6]. G. Li, Y. Shi and A. Javadi-Abhari, “Software-Hardware Co-Optimization for Computational Chemistry on Superconducting Quantum Processors,” 2021 ACM/IEEE 48th Annual International Symposium on Computer Architecture (ISCA), Valencia, Spain, 2021, pp. 832-845, doi: 10.1109/ISCA52012.2021.00070
[7]. Cornell, W., Cieplak, P., Bayly, C., Gould, I., Merz, K., Ferguson, D., Spellmeyer, D., Fox, T., Caldwell, J. & Kollman, P. (1995). A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. Journal of the American Chemical Society, 117, 5179—5197
[8]. Liebl, K., & Zacharias, M. (2021). Tumuc1: A New Accurate DNA Force Field Consistent with High-Level Quantum Chemistry. Journal of Chemical Theory and Computation, 17(11), 7096-7105. https://doi.org/10.1021/acs.jctc.1c00682
[9]. Brooks, B. R., Brooks, C. L., 3rd, Mackerell, A. D., Jr, Nilsson, L., Petrella, R. J., Roux, B., Won, Y., Archontis, G., Bartels, C., Boresch, S., Caflisch, A., Caves, L., Cui, Q., Dinner, A. R., Feig, M., Fischer, S., Gao, J., Hodoscek, M., Im, W., Kuczera, K., … Karplus, M. (2009). CHARMM: the biomolecular simulation program. Journal of computational chemistry, 30(10), 1545–1614. https://doi.org/10.1002/jcc.21287
[10]. MacKerell Jr, A. D., Bashford, D., Bellott, M. L. D. R., Dunbrack Jr, R. L., Evanseck, J. D., Field, M. J., ... & Karplus, M. (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. The journal of physical chemistry B, 102(18), 3586-3616.
[11]. Case, D. A., Cheatham, T. E., 3rd, Darden, T., Gohlke, H., Luo, R., Merz, K. M., Jr, Onufriev, A., Simmerling, C., Wang, B., & Woods, R. J. (2005). The Amber biomolecular simulation programs. Journal of computational chemistry, 26(16), 1668–1688. https://doi.org/10.1002/jcc.20290
[12]. Cornell, W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, K. M., Ferguson, D. M., ... & Kollman, P. A. (1995). A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. Journal of the American Chemical Society, 117(19), 5179-5197
[13]. Nester, K., Gaweda, K., & Plazinski, W. (2019). A GROMOS Force Field for Furanose-Based Carbohydrates. Journal of chemical theory and computation, 15(2), 1168–1186. https://doi.org/10.1021/acs.jctc.8b00838
[14]. Ditler, E, Luber, S. Vibrational spectroscopy by means of first-principles molecular dynamics simulations. WIREs Comput Mol Sci. 2022; 12:e1605. https://doi.org/10.1002/wcms.1605
[15]. Ponder, J. W., & Case, D. A. (2003). Force fields for protein simulations. Advances in protein chemistry, 66, 27-85
[16]. He, X., Liu, S., Lee, T. S., Ji, B., Man, V. H., York, D. M., & Wang, J. (2020). Fast, accurate, and reliable protocols for routine calculations of protein–ligand binding affinities in drug design projects using AMBER GPU-TI with ff14SB/GAFF. ACS omega, 5(9), 4611-4619
[17]. Dong, X., Yuan, X., Song, Z., & Wang, Q. (2021). The development of an Amber-compatible organosilane force field for drug-like small molecules. Physical Chemistry Chemical Physics, 23(22), 12582-12591
[18]. Ngo, S. T., Nguyen, M. T., & Nguyen, M. T. (2017). Determination of the absolute binding free energies of HIV-1 protease inhibitors using non-equilibrium molecular dynamics simulations. Chemical Physics Letters, 676, 12-17. DOI: 10.1016/j.cplett.2017.03.034
[19]. Lins, R. D., & Hünenberger, P. H. (2005). A new GROMOS force field for hexopyranose‐based carbohydrates. Journal of computational chemistry, 26(13), 1400-1412
[20]. de Arenaza, I. M., Meaurio, E., & Sarasua, J. R. (2012). Analysis of the miscibility of polymer blends through molecular dynamics simulations. Polymerization
[21]. He, M., Qiu, F., & Lin, Z. (2013). Towards high-performance polymer-based thermoelectric materials. Energy & Environmental Science, 6(5), 1352-1361
[22]. Gartner III, T. E., & Jayaraman, A. (2019). Modeling and Simulations of Polymers: A Roadmap. Macromolecules, 52(3), 755-786. DOI: 10.1021/acs.macromol.8b01836
[23]. Maryasin, B., Marquetand, P., & Maulide, N. (2018). Machine learning for organic synthesis: are robots replacing chemists?. Angewandte Chemie International Edition, 57(24), 6978-6980
[24]. Emmert-Streib, F. (2021). Grand Challenges for Artificial Intelligence in Molecular Medicine. Frontiers in Molecular Medicine, 1. DOI: 10.3389/fmmed.2021.734659
[25]. Kim, H., Kim, E., Lee, I., Bae, B., Park, M., & Nam, H. (2020). Artificial intelligence in drug discovery: a comprehensive review of data-driven and machine learning approaches. Biotechnology and Bioprocess Engineering, 25, 895-930
[26]. Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O., & Walsh, A. (2018). Machine learning for molecular and materials science. Nature, 559(7715), 547-555
[27]. Zubarev, D., Mendes, C. R., Brazil, E. V., Cerqueira, R., Schmidt, K., Segura, V., ... & Sanders, D. (2022). Toward Human-AI Co-creation to Accelerate Material Discovery. arXiv preprint arXiv:2211.04257
[28]. Gomes, C.P., Selman, B. & Gregoire, J.M. Artificial intelligence for materials discovery.MRS Bulletin 44, 538–544 (2019). DOI: 10.1557/mrs.2019.158
[29]. Kailkhura, B., Gallagher, B., Kim, S., Hiszpanski, A., & Han, T. Y. J. (2019). Reliable and explainable machine-learning methods for accelerated material discovery. npj Computational Materials, 5(1), 108
[30]. Qayyum, F., Kim, D. H., Bong, S. J., Chi, S. Y., & Choi, Y. H. (2022). A Survey of Datasets, Preprocessing, Modeling Mechanisms, and Simulation Tools Based on AI for Material Analysis and Discovery. Materials, 15(4), 1428
[31]. Zylla, J. L., Gnimpieba, E., Bomgni, A. B., Sani, R. K., Subramaniam, M., Lushbough, C., ... & Chundi, P. (2023). Convergence research and training in computational bioengineering: a case study on AI/ML driven biofilm-material interaction discovery
[32]. Zhang, L., Han, J., Wang, H., Car, R., & Weinan, E. J. P. R. L. (2018). Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Physical review letters, 120(14), 143001
[33]. Zhang, L., Huang, K., Yang, Y. I., & Shi, L. (2022). Combined artificial intelligence and molecular dynamics (AI-MD) methods. Frontiers in molecular biosciences, 9, 1012785
[34]. Ye, F., Liang, Z., & Broussy, Y. (Eds.). (2022). Computer-aided drug design: Drug discovery, computational modelling, and artificial intelligence