







1. Introduction
Lung cancer has been one of the major causes of cancer death, reporting 1.8 million cases in 2020 [1]. NSCLC accounts for 80%–85% of all lung cancer cases [2]. Genetic anomalies in the tyrosine kinase domain of EGFR are one of the main factors contributing to the development of NSCLC [3]. EGFR tyrosine kinase (EGFR-TK) is therefore a key target for the therapy of NSCLC.
Advanced NSCLC with EGFR activating mutations have been treated with EGFR tyrosine kinase inhibitor (EGFR-TKI) drugs. The representative drugs are gefitinib, erlotinib, and afatinib [4-7]. The first-generation drugs are non-selective, reversible inhibitors. They contain the structural motif of quinolinamine. Erlotinib and gefitinib are two representative medications in this class. These drugs considerably slow the progress of the disease compared to conventional chemotherapy medications, however, when using these class inhibitors, EGFRT790M mutations result in developing therapeutic resistance.
The second-generation of EGFR-TKI drugs, which are non-selective, irreversible inhibitors, were required due to the quick onset of medication resistance. They also employ the structural motif of quinolinamine. The two medications that serve as examples are afatinib and dacomitinib. The majority of second-generation inhibitors include a unique functional class of Michael addition receptors, such as unsaturated amide. The sulfhydryl group of Cys797 can be covalently bound by these unsaturated amides to create irreversible inhibition, boosting the affinity with EGFR-TK proteins [8]. Selectivity is lacking in the second-generation drugs, with both the mutant EGFR and wild-type inhibited by them. In the skin and gastrointestinal tract, the absence of selectivity over normal EGFR causes adverse effects like skin rashes and diarrhea [9]. The medication resistance brought on by the T790M mutation was also not considerably reduced by the second-generation drugs.
The need for third-generation irreversible EGFR-TKI drugs is driven by medication resistance. Third- generation drugs have pyrimidine as their main structural component. The T790M mutation and the mutations (exon 19 deletion mutation and L858R mutation) are specifically targeted by these inhibitors by binding to the C797 position [10]. The representative medication is osimertinib [11]. In this paper, we describe the medicinal chemistry work to discover osimertinib (AZD9291), a potent inhibitor of the mutant forms while with a good selectivity over the wild-type form.
The strategy of osimertinib synthesis and production has different focuses at different drug development stages. At the early phase of drug discovery, medicinal chemists focused more on finding a good drug candidate through lead optimization and focused less on process development including synthetical route development and optimization. Finlay et al. described how osimertinib was first synthesized [12]. The synthetic approach includes seven steps, though, and a low overall yield. This inspired scientists to find more efficient synthetic pathways. Especially at the commercial product production stage, efforts need to be made to bring the cost of goods down so that patients can benefit from an affordable low drug price. In this paper, the chemical reactions to produce osimertinib, both from medicinal chemistry approach and from industrial process chemistry approach, are also discussed.
2. Results and Discussion
2.1. Development of EGFR-TKI drugs for the treatment of NSCLC
EGFR is a tyrosine kinase and a transmembrane protein. Extracellular EGF binding domain, transmembrane domain, and cytoplasmic domain are the three components that make up EGFR. The protein tyrosine kinase (PTK) domain and the C-terminal phosphorylation domain are the two components of the cytoplasmic domain. Epidermal growth factor (EGF) ligands will start a downstream signaling cascade when they bind to EGFR [13]. In Figure 1, the mechanism is displayed.
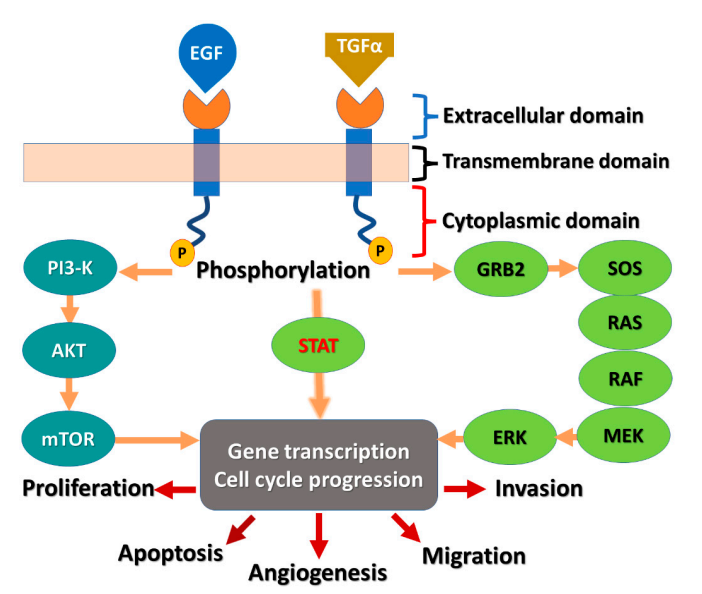
Figure 1. EGFR receptor and the signaling pathway.
Many malignancies are linked to the overexpression of the EGFR. The pro-activation of downstream signaling pathways that fuel pro-proliferative and survival cellular signals is facilitated by ligand-induced activation of EGFR. Exon 19 deletion and L858R are two activating mutations that have been found in EGFR in NSCLC patients. The receptor becomes generally active as a result, unaffected by ligand activation. This causes the signaling pathways downstream of EGFR to persistently become hyperactive. Small-molecule kinase inhibitors are therefore used to inhibit activated EGFR in order to stop its signaling and lessen its carcinogenic potential. In the end, it prevents the growth of tumors.
In 2003, FDA approved the first EGFR-TKI drug, gefitinib. In 2004, FDA approved erlotinib. The PTK domain of the EGFR is reversibly bound by medications in this class. They bind to ATP, which suppresses EGFR activation and cellular growth. A quinazoline nucleus bonds to a substituted aniline moiety in the chemical structures of the medicines. Patients with NSCLC who have activating mutations in EGFR are treated with first-generation drugs like gefitinib and erlotinib. However, within 10–16 months, over 70% of patients who initially respond acquire resistance. The drug resistance is linked to the acquisition of a secondary mutation called the gatekeeper mutation. This T790M mutation occurs in EGFR exon 20, along with an activating mutation (shown in Figure 2) [12].
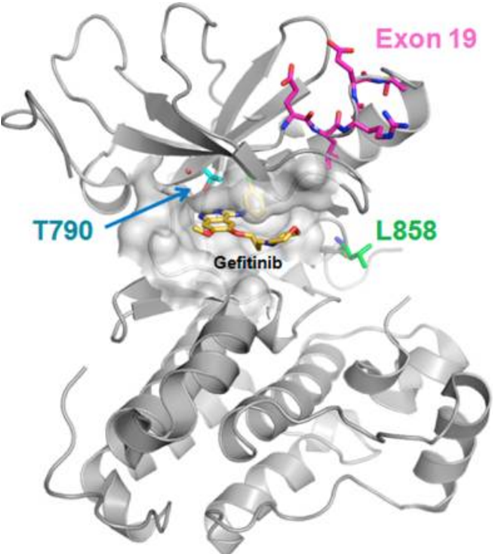
Figure 2. Activating and resistant mutations in the kinase domain.
Advanced NSCLC patients whose tumor activating mutations occur in deletions in exons 19 and 21 are found to respond best to first-generation drugs like gefitinib and erlotinib. Despite the fact that patients with EGFRm+ malignancies often respond well to first-generation drugs at first, the majority of patients develop drug resistance after 9 to 14 months of therapy. In addition, suppression of wild-type EGFR in the skin and gastrointestinal tract leads to adverse effects such as diarrhea and skin rashes. A second mutation in the EGFR converts the amino acid 790 from threonine to methionine (T790M). The T790M mutation causes steric hindrance and enhanced ATP affinity, which renders the receptor resistant to inhibition. Second-generation drugs were therefore developed. Afatinib and dacomitinib are two representative drugs. FDA approved afatinib in 2013 and dacomitinib in 2018. These medications have a Michael acceptor site that enables covalent bond to the EGFR. It has an advantage over the first-generation of drugs and causes the irreversible inhibition of the kinase activity. These medications' chemical structures contain a quinazoline or quinoline nucleus, which enables them to contain a crotonamide side chain as a Michael acceptor site. However, in tissues or organs like the skin and gastrointestinal system, they lack selectivity over wild-type EGFR, leading to adverse effects including skin rash and diarrhea. Furthermore, they are unable to overcome patients' T790M-mediated resistance. A drug that can more successfully target T790M cancers without harming wild-type EGFR is thus needed. Targeting T790M and sensitizing mutations while not hitting wild-type EGFR is the goal of the third-generation drugs.
As a third-generation drug, AZD9291 (osimertinib) was discovered and developed. It is a mono-anilino-pyrimidine compound. By targeting the cysteine-797 residue in the ATP binding site, it covalently binds to the EGFR kinase in an irreversible manner [14]. A pyrimidine nucleus is bonded to a substituted anilino or phenoxy moiety in the chemical structure. As seen in Figure 3, these molecules have an acrylamide group that can bond covalently with the cysteine residue of the EGFR.
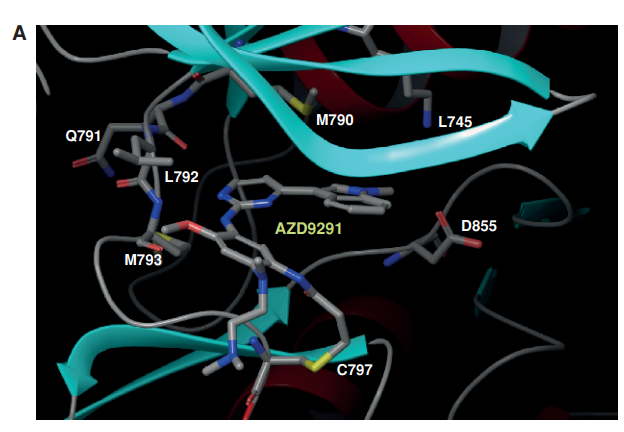

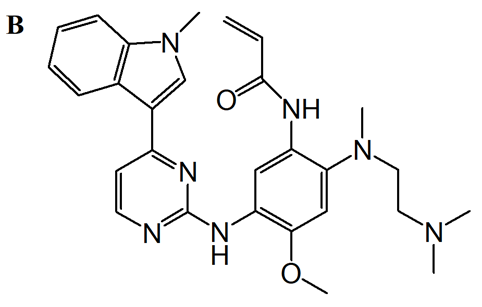
Figure 3. AZD9291 binding mode and structure. A: covalently binds to EGFRT790M through Cys797 via the acrylamide group; indole group next to the T790 residue; position the amine moiety in the solvent channel. The pyrimidine core creates two hydrogen bonds to the hinge region (Met793). B: chemical structure of AZD9291.
The development of three generations of EGFR-TKIs are shown below in Figure 4 [15]. It shows the drug resistant mechanisms, the evolution of different generations of drugs and their representative drugs.
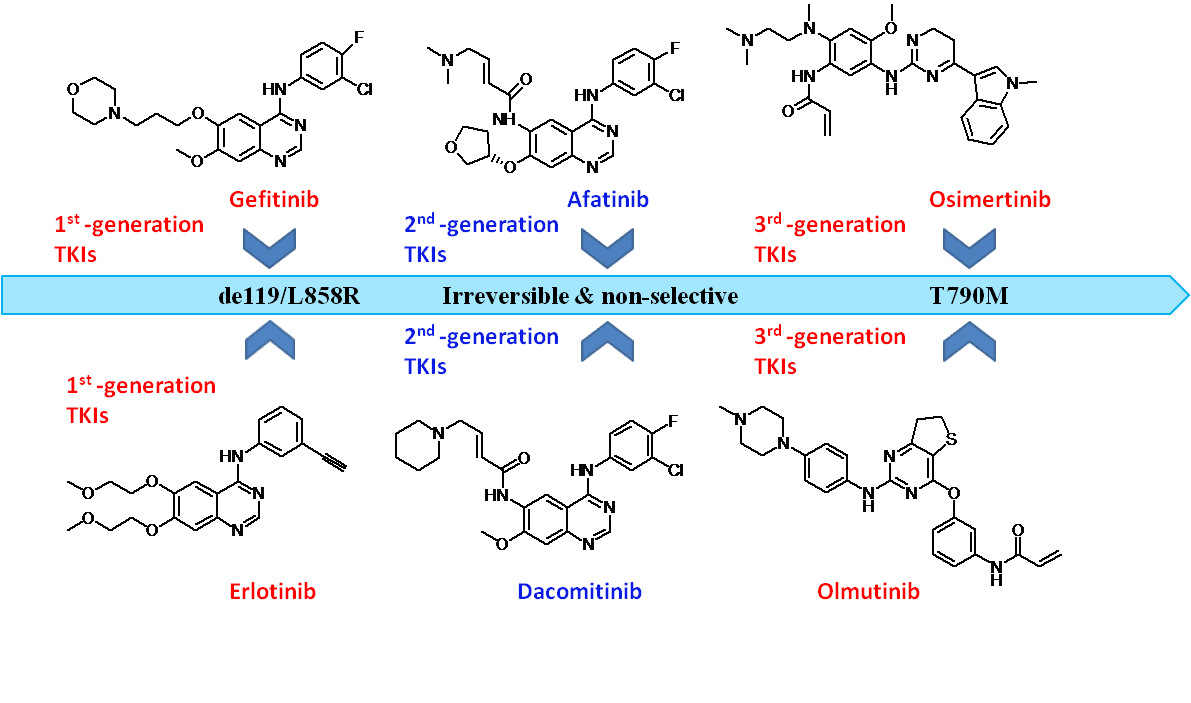
Figure 4. Development of three generations of EGFR-TKIs.
2.2. Lead optimization
Lead compounds were optimized to have the inhibition of double mutants but not hitting the wild type. In Table 1, activity profile of market approved EGFR Inhibitors is shown. These activities included the wild-type enzyme (WT), the exon 19 deletion activating mutation (AM), and the L858R/T790M double-mutant enzyme (DM) [12].
Table 1. Cell Base Activity Profile.
Drug | T790M/L858R Double Mutant (M) | Exon 19 Deletion Activating Mutant (M) | Wild-Type Form (M) | Margin of Double Mutant/Wild Type |
Gefitinib (1st generation) | 3.3 | 0.0087 | 0.062 | 0.02 |
Erlotinib (1st generation) | 7.6 | 0.0059 | 0.077 | 0.01 |
Afatinib (2nd generation) | 0.023 | 0.00057 | 0.012 | 0.53 |
Dacomitinib (2nd generation) | 0.042 | 0.00063 | 0.011 | 0.27 |
In Table 1, the smaller the numeric number shown in the second, third, and forth columns, the stronger the activity and inhibition on the wild-type form or the mutant form, and consequently the more potent of the drug. For DM/WT margin, the larger the better, for we don’t want to hit wild type. Gefitinib and erlotinib are active against the activating mutant and more active against the wild-type EGFR. However, they decreased (3.3 and 7.6 M, respectively) cell base activity against double-mutant EGFR. In comparison to gefitinib and erlotinib, afatinib and dacomitinib are more potent against wildtype, activating-mutant, and double mutant. However, the selectivity against the double mutant and against wild-type EGFR has a small margin. (i.e., margin of <1).
The wild-type and double mutant enzymes were used to assess a group of chemicals to find mutant-selective pyrimidine core scaffolds. When compared to wild-type EGFR, these drugs were selectively active against double mutant EGFR.A representative compound template is shown in Figure 5.
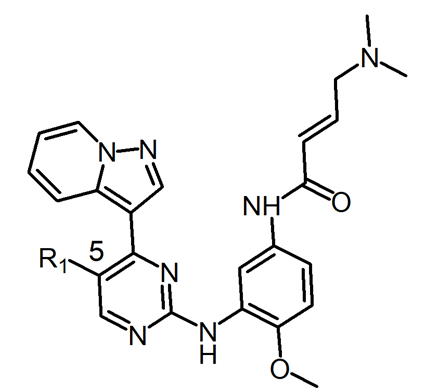
Figure 5. The lead compound template for lead optimization to discover drug candidates.
The 5-position of the compound template was positioned next to the gatekeeper residue. The potency and selectivity of drugs are impacted by changing the substitutes in this position. Chlorine (compound 1), which is relatively lipophilic, was discovered as the first hit. By changing this group, it was possible to assess how the compound's lipophilicity affected its potency and selectivity.
Table 2. Structure and Cell Base Data.
Compound# | Substituent R1 | Activating Mutant (μM) | Double Mutant (μM) | Wild Type (μM) | Margin of Double Mutant/Wild Type | lipophilicity logD7.4 |
1 | Chlorine | 0.40 | 0.096 | 23 | 237 | 3.6 |
2 | Hydrogen | 0.39 | 0.25 | 20 | 80 | 2.6 |
3 | Florine | 0.38 | 0.22 | 19 | 85 | 3.3 |
4 | Methyl | 0.25 | 0.29 | 24 | 82 | 3 |
5 | S(=O)Me | 1.3 | 15.2 | >30 | >2 | 1.3 |
Compound 2 decreased the double-mutant activity in comparison to the hit compound 1 (0.25 vs 0.096 M), whereas compound 2 had no effect on the activating-mutant activity (0.39 vs 0.40 M). Compound 2 increased chlorine's interaction with the methionine gatekeeper residue, decreasing the double-mutant selectivity from 237 to 80-fold. However, the information gathered about these compounds revealed that preserving a good wild-type selectivity while eliminating chlorine from the 5-position could lead to reasonable potency on the activating and double mutants. The overall lipophilicity of these compounds may decrease if the chlorine is removed. Compound 1 is selected as the lead compound for further evaluation.
In addition to compound 1, compounds 6 and 7 are also identified as lead compounds, as shown in Figure 6 [16]. Compound 6 showed broadly similar double mutant potency to 1. It increases activating mutant activity while decreasing IGF1R activity (IGF1R operates as a cancer-promoting factor). Compound 7 exhibits good selectivity for the receptor mutant variants. In both the double mutant ATP binding site and the IGF1R, a lipophilic group at the 5-position presents an interaction with the methionine. Kinetic selectivity may be achieved by irreversible binding to the double mutant.
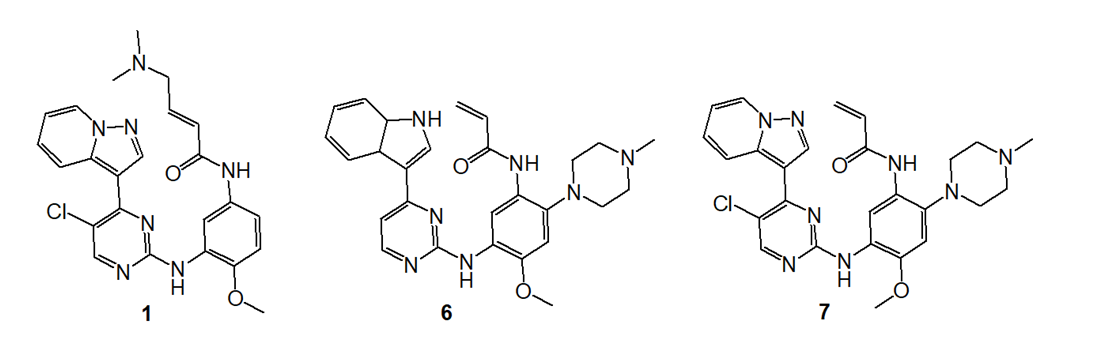
Figure 6. Structures of Lead Optimization Start Points.
Osimertinib was discovered and selected as a drug by lead optimization from lead compounds, 1, 6, and 7, in specific, through medicinal chemistry efforts of lead compound whose structure is shown in Figure 7 for potency (against EGFR mutant types), selectivity (between wild type and mutant type), drug like properties (such as solubility, lipophilicity), and good pharmacokinetic (such as oral bioavailability) and safety profile (Table 3) [16]. Osimertinib was further developed to become a market approved drug.
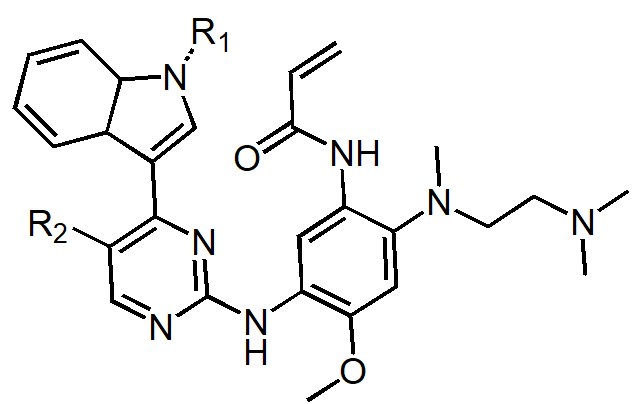
Figure 7. Structure of the lead of compound that discover osimertinib.
Table 3. Structure and Cell Base Data.
Compound Number 8 | Compound Number 9 | Compound Number 10 | Compound Number 11 | Compound Number 12 Osimertinib | |
R1 Substituent, R2 Substituent | CH3, CN | CH3, Cl | H, Cl | CH3, CH3 | CH3, H |
Double mutant IC50 (nM) | 0.9 | 2 | 0.2 | 1 | 15 |
Activating mutant IC50 (nM) | 1 | 2 | 0.6 | 2 | 17 |
Wild type IC50 (nM) | 46 | 58 | 11 | 71 | 480 |
IGF1R Assay IC50 (nM) | 38 | 40 | 7 | 196 | 2900 |
hERG Assay IC50 (M) | 4.3 | 7.4 | 14.8 | 15.6 | 16.2 |
Lipophilicity logD7.4 | 2.7 | 3.3 | 3.3 | 2.8 | 3.4 |
Solubility at pH 7.4 (M) | 57 | 759 | 75 | 576 | 7 |
Rat oral bioavailability F (%) | 6 | 7 | 1 | 45 |
High cellular potency can be achieved by incorporating the basic side chains (Table 3). The substituent functional group of the pyrimidine ring at the 5-position influences double mutant potency, IGF1R potency, and hERG binding. A 5-substituent improves the efficacy of double mutants via interacting with the methionine gatekeeper residue. This kinase's methionine gatekeeper is responsible for a comparable trend in IGF1R potency. Furthermore, the receptor's potency against the activating mutant and wild-type versions was improved. Compared to earlier EGFR TKI agents, compound 12 (osimertinib) shows higher potency and better selectivity to the wild-type form.
2.3. Synthesis:
Osimertinib was synthesized by a variety of synthetic routes. The original and widely used technique creates 15 via the reaction of Grignard reagent and halide, as outlined in Figure 8.
2.3.1. Medicinal chemistry route [16]. The initial seven-step process (Figure8) for making osimertinib: react 13 with MeMgBr, followed by the addition of 14, which results in 15 in a yield of 46%. NaH/MeI methylation yields 96% of the indole 16 product. After crystallization, 18 is created by substituting aniline for the other pyrimidine chloride under acidic conditions at 85°C, yielding 85% of the product. Under microwave conditions, a SNAr reaction with the diamine 19 results in 20 in a 62% yield. Penultimate intermediate 21 is produced in 85% by reduction. Osimertinib is produced through acylation in 39% of yields.
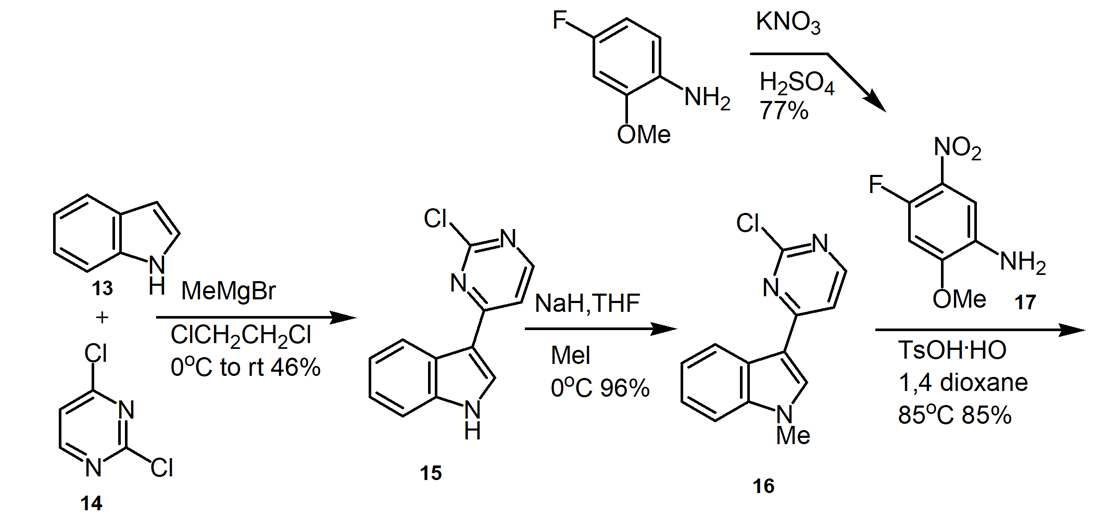
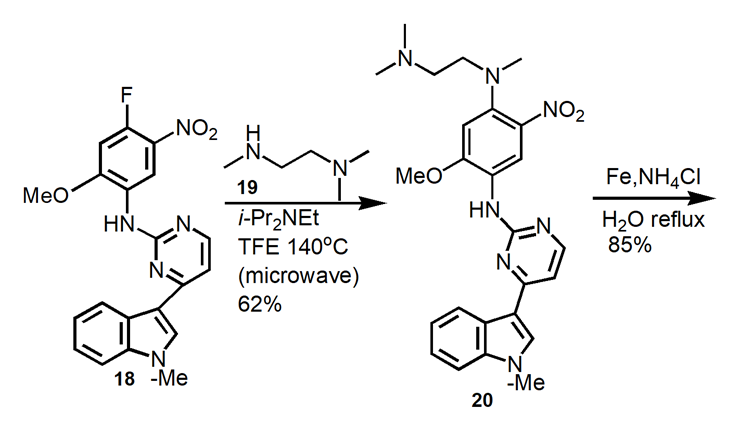
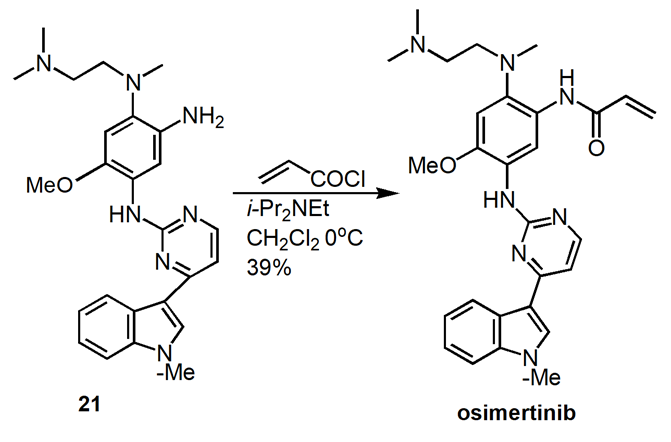
Figure 8. Medicinal Chemistry Route to Osimertinib.
In this medical chemistry route, hundred milligram scale of osimertinib was produced in 8.6% yield over seven reaction steps. Column chromatography purification of products was applied. The synthesis route and process are not friendly for scale up. The drawbacks of this synthetic route include: (1) use of microwave condition which is not a conventional technique for commercial product production, (2) the overall yield is very low.
2.3.2. Process improvement route. Figure 9 shows an outline and summary of an improved process from the medicinal chemistry route.
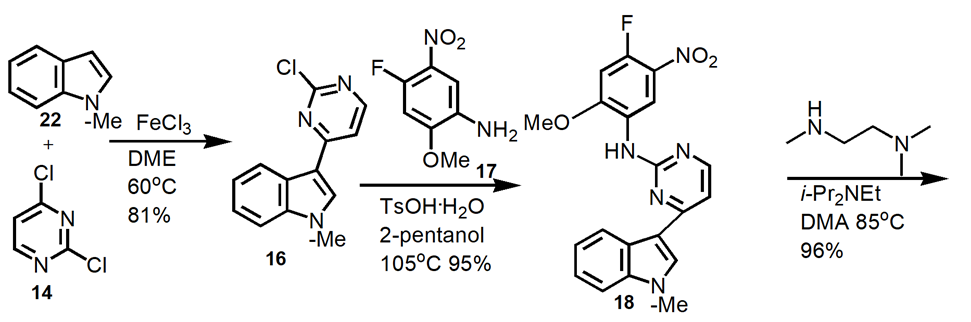
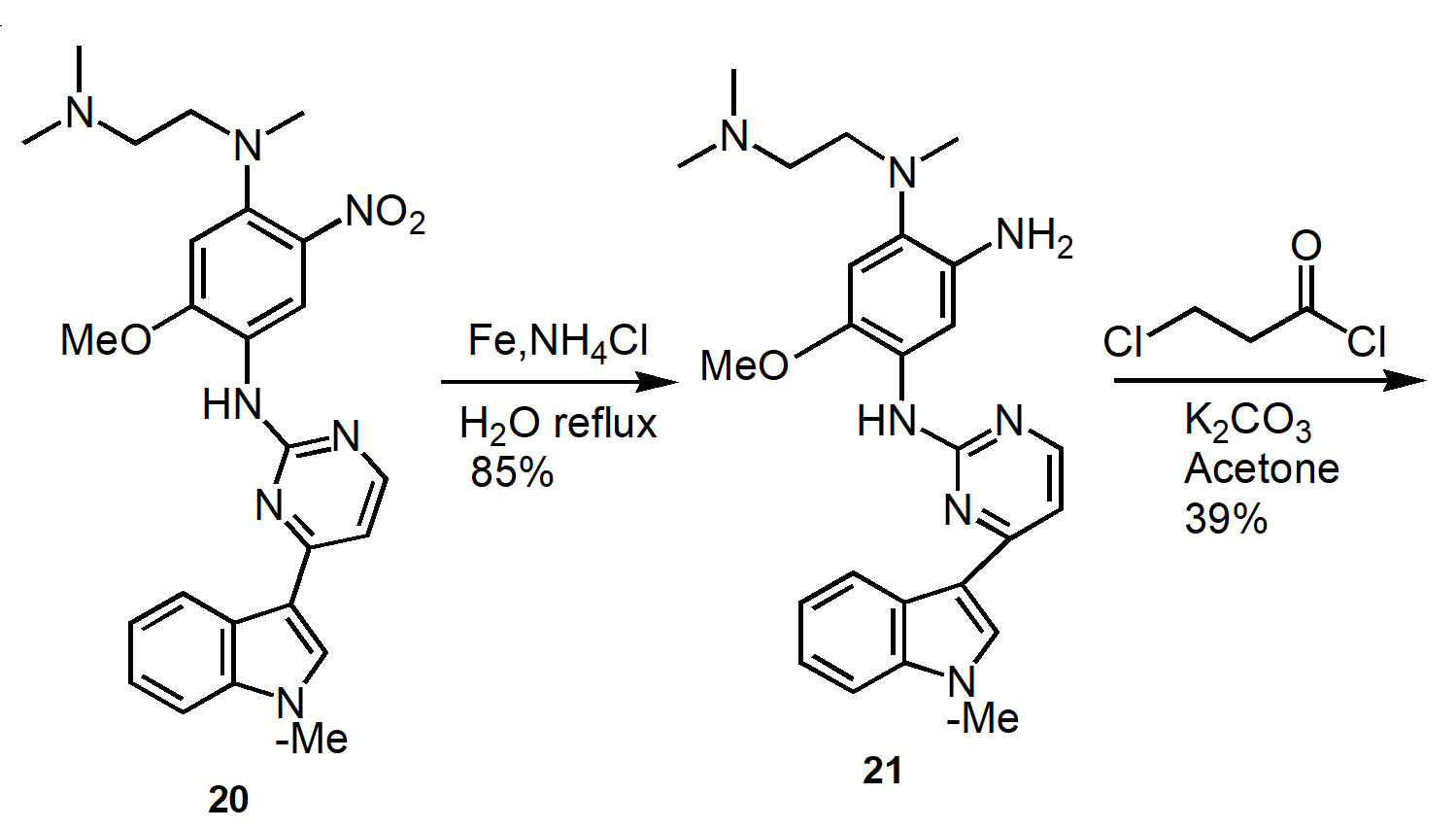
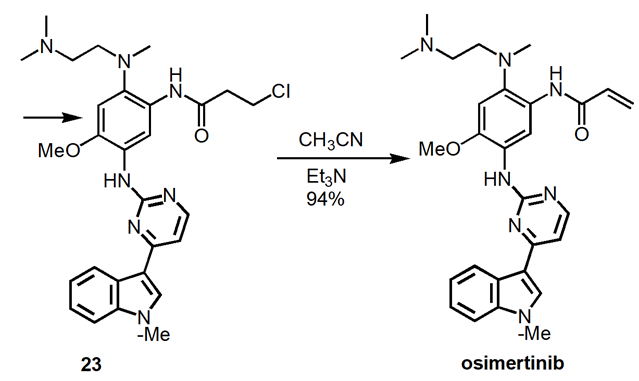
Figure 9. Improved Manufacturing Route to Osimertinib.
Produce intermediate 16 at 60oC in 81% yield. React 16 with 17 at 105oC to produce 18 in 95% yield. Convert 18 to 20 in 96% yield at 85oC. Reduction of 20 to produce 21 in 85% yield. Amidation of 21 to form 23 in 95% yield. Elimination of 23 to produce osimertinib in 94% yield.
The apparent manufacturing process optimizes the medicinal chemistry route. Non-conventional methods such as chromatography and microwave heating were removed. Replace environmentally undesirable solvents with environmentally friendly solvents.
2.3.3. Alternate Routes to Osimertinib. The identical bond disconnections occur along a different order along a different path to osimertinib (Figure 10) [17]. Construct the two complex intermediates, 16 and 27 and then couple in the final step. The advantage of this synthetic route is an improvement in the overall reaction yield to 65% for the 6 steps.
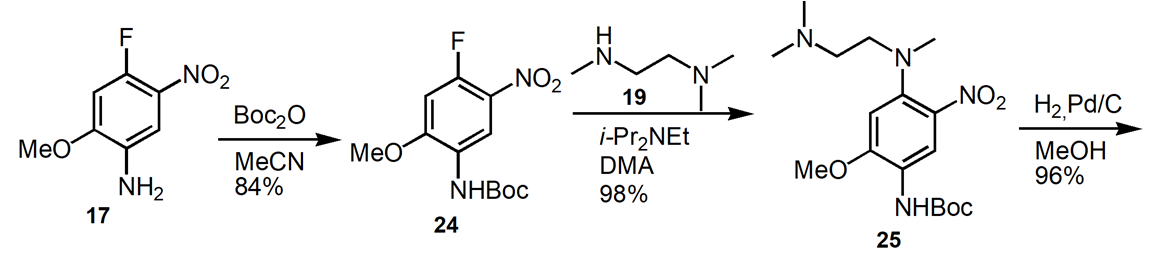
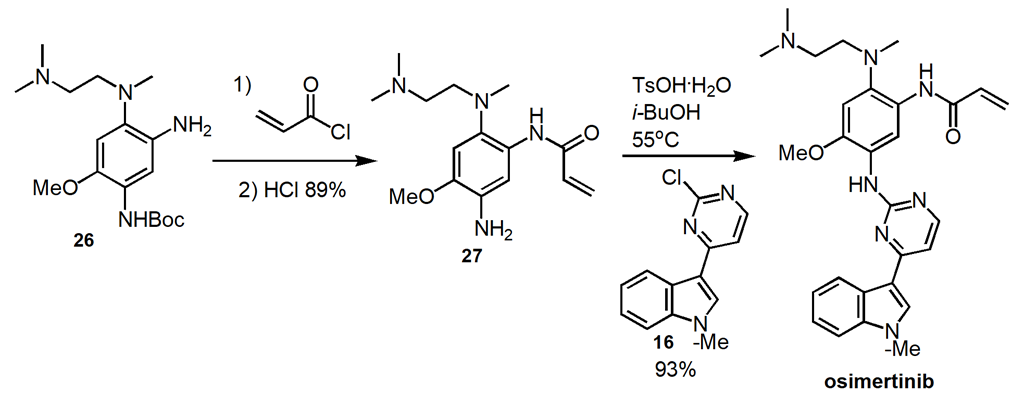
Figure 10. Alternate Route to Osimertinib.
2.3.4. A commercial consideration route. As shown in Figure 11, this synthetic route uses the commercially available materials. The reaction steps include nitration, guanidination, methylation, and catalytic hydrogenation. A 40.4% yield of the final product over six steps and 99.1% purity are achieved [18].
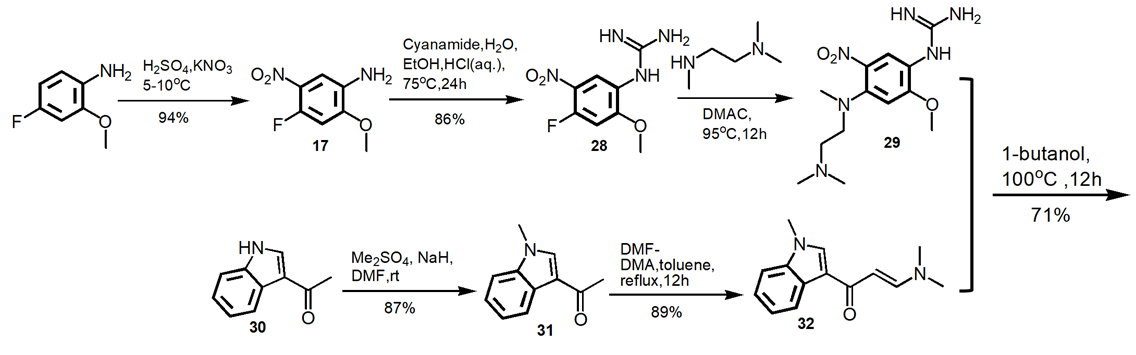
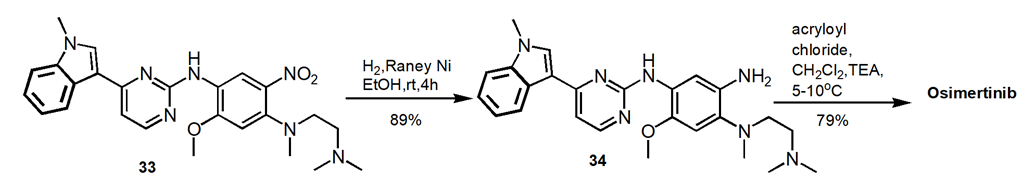
Figure 11. Alternate Route to Osimertinib.
3. Conclusion
We have described the three generations of EGFR-TKIs for the treatment of non-small cell lung cancer, while illustrating a typical drug discovery medicinal chemistry approach to discover a new drug from lead optimization to clinical candidate nomination of AZD9291 (osimertinib). Osimertinib overcomes on-target resistance to Gefitinib and Erlotinib of the first-generation drugs while improving the drug selectivity issue from second-generation drugs of Afatinib and Dacomitinib. Osimertinib targets both sensitizing (or activating) and the T790M+ mutant EGFR in an irreversible and selective manner, with reduced activity toward the wild-type EGFR, enabling all patients to benefit from safety and improved survival. Four synthetic routes to produce osimertinib, each of which serves for different purposes at different drug development stages, are also compared: from the early phase of drug discovery focusing medicinal chemistry lead optimization to the late phase of drug development focusing on cost effective commercial product. By adopting the commercially available starting materials, minimizing the synthetic reaction steps (e.g. from 7 steps to 6 steps), using more conventional reaction conditions (e.g. eliminate microwave reaction condition), and increasing the overall reaction yield (e.g. from 8.6% to 40.4%), the cost of goods is decreased so that patients can spend less money on medicine and enjoy a more affordable drug.
References
[1]. World Health Organization. Cancer Fact Sheet 2017, Available at: http://www.who.int/mediacentre/factsheets/fs297/en/. Accessed: January 7, 2020.
[2]. Thai, A. A., Solomon, B. J., Sequist, L. V., Gainor, J. F., Heist, R.S. (2021)Lung cancer, Lancet, 398: 535–554.
[3]. Sharma, S. V., Bell, D. W., Settleman, J., Haber, D. A. (2007) Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer, 7: 169-181.
[4]. Mok, T. S., Wu, Y.L., Thongprasert, S., Yang, C.H., Chu, D.T., Saijo, N.,Sunpaweravong, P., Han, B., Margono, B., Ichinose, Y., Nishiwaki, Y., Ohe, Y., Yang, J.J.,Chewaskulyong, B., Jiang, H., Duffield, E. L., Watkins, C. L.,Armour, A. A., Fukuoka, M. (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med., 361: 947−957.
[5]. Shepherd, F. A., Pereira, J. R.,Ciuleanu, T., Tan, E. H., Hirsh, V.,Thongprasert, S., Campos, D.,Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; Kooten, M. V.; Dediu, M.; Findlay, B.; Tu, D.; Johnston, D., Bezjak, A., Clark, G., Santabarbara, P., Seymour, L. (2005) Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med., 353: 123−132.
[6]. Cataldo, V. D., Gibbons, D. L., Perez-Soler, R., Quinta ́ s-́Cardama, A. (2011) Treatment of non−small-cell lung cancer with erlotinib or gefitinib. N. Engl. J. Med., 364: 947−955.
[7]. Wu, Y.-L., Zhou, C., Hu, C.-P., Feng, J., Lu, S., Huang, Y., Li, W., Hou, M., Shi, J. H., Lee, K.-Y., Xu, C. R., Massey, D., Kim, M., Shi, Y., Geater, S. L. (2014) Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol., 15: 213−222.
[8]. Hoffknecht, P.,Tufman, A., Wehler, T., Pelzer, T., Wiewrodt, R., Schütz, M., Serke, M., Stohlmacher-Williams, J., Marten, A., Maria Huber, R., Dickgreber, N. J. (2015) Efficacy of the irreversible ErbB family blocker Afatinib in epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI)–Pretreated non–small-cell lung cancer patients with brain metastases or leptomeningeal disease, J. Thorac. Oncol., 10: 156–163.
[9]. Westover, D., Zugazagoitia, J., Cho, B. C., Lovly, C. M., Paz-Ares, L. (2018) Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors, Ann. Oncol., 29: i10–i19.
[10]. Kim, Y., Ko, J., Cui, Z., Abolhoda, A., Ahn, J. S., Ou, S. H., Ahn, M. J., Park, K. (2012)The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor, Mol. Cancer Ther., 11: 784–791.
[11]. Liam, K. (2017) Osimertinib as first-line treatment of EGFR mutant advanced nonsmall cell lung cancer, Lung Cancer Res., 6: S62–S66.
[12]. Ward, R. A., Anderton, M. J., Ashton, S., Bethel, P. A., Box, M., Butterworth, S., Colclough, N., Chorley, C. G.,Chuaqui, C., Cross, D. A., Dakin, L. A., Debreczeni, J. E., Eberlein, C., Finlay, M. R. V., Hill, G. B., Grist, M.,Klinowska, T. C., Lane, C., Martin, S., Orme, J. P., Smith, P., Wang, F., Waring, M. J. (2013) Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J. Med. Chem., 56: 7025−7048.
[13]. Abourehab, M. A. S., Alqahtani, A. M., Youssif, B. G. M.,Gouda, A. M. (2021) Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules, 26: 6677
[14]. Cross, D. A. E., Ashton, S.,Ghiorghiu, S., Eberlein, C.,Nebhan, C.,Spitzler, P., Orme, J., Finlay, M. R. V., Ward, R. A., Mellor, M., Hughes, G., Rahi, A., Jacobs, V., Red Brewer, M., Ichihara, E., Sun, J., Jin, H., Ballard, P., Al-Kadhimi, K., Rowlinson, R.,Klinowska, T., Richmond, G.,Cantarini, M., Kim, D.-W., Ranson, M., Pao, W. (2014) AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery, 4: 1046-1061
[15]. Xu, L., Xu, B., Wang, J., Gao, Y., He, X., Xie, T., Ye, X. Y. (2023) Recent advances of novel fourth generation EGFR inhibitors in overcoming C797S mutation of lung cancer therapy, Eur. J. Med. Chem., 245: 114900
[16]. Finlay, M.R.V., Anderton, M., Ashton, S., Ballard, P., Bethel, P.A., Box, M.R., Bradbury, R.H., Brown, S.J., Butterworth, S., Campbell, A. (2014) Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem., 57: 8249–8267.
[17]. Liu, H.,Lv, Y., Li, Y., Cai, J., Chen, J., Qin, Y., Ji, M.(2015)J. Chem. Res., 39: 318-320.
[18]. Zhu, G., Wang, X., Wang, F., Mao, Y., Wang, H. (2017) New and Convergent Synthesis of Osimertinib. J. Heterocycl. Chem., 54: 2898–2901.
Cite this article
Li,V. (2024). The discovery of osimertinib (AZD9291) for the treatment of lung cancer and its chemistry. Theoretical and Natural Science,35,225-236.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. World Health Organization. Cancer Fact Sheet 2017, Available at: http://www.who.int/mediacentre/factsheets/fs297/en/. Accessed: January 7, 2020.
[2]. Thai, A. A., Solomon, B. J., Sequist, L. V., Gainor, J. F., Heist, R.S. (2021)Lung cancer, Lancet, 398: 535–554.
[3]. Sharma, S. V., Bell, D. W., Settleman, J., Haber, D. A. (2007) Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer, 7: 169-181.
[4]. Mok, T. S., Wu, Y.L., Thongprasert, S., Yang, C.H., Chu, D.T., Saijo, N.,Sunpaweravong, P., Han, B., Margono, B., Ichinose, Y., Nishiwaki, Y., Ohe, Y., Yang, J.J.,Chewaskulyong, B., Jiang, H., Duffield, E. L., Watkins, C. L.,Armour, A. A., Fukuoka, M. (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med., 361: 947−957.
[5]. Shepherd, F. A., Pereira, J. R.,Ciuleanu, T., Tan, E. H., Hirsh, V.,Thongprasert, S., Campos, D.,Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; Kooten, M. V.; Dediu, M.; Findlay, B.; Tu, D.; Johnston, D., Bezjak, A., Clark, G., Santabarbara, P., Seymour, L. (2005) Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med., 353: 123−132.
[6]. Cataldo, V. D., Gibbons, D. L., Perez-Soler, R., Quinta ́ s-́Cardama, A. (2011) Treatment of non−small-cell lung cancer with erlotinib or gefitinib. N. Engl. J. Med., 364: 947−955.
[7]. Wu, Y.-L., Zhou, C., Hu, C.-P., Feng, J., Lu, S., Huang, Y., Li, W., Hou, M., Shi, J. H., Lee, K.-Y., Xu, C. R., Massey, D., Kim, M., Shi, Y., Geater, S. L. (2014) Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol., 15: 213−222.
[8]. Hoffknecht, P.,Tufman, A., Wehler, T., Pelzer, T., Wiewrodt, R., Schütz, M., Serke, M., Stohlmacher-Williams, J., Marten, A., Maria Huber, R., Dickgreber, N. J. (2015) Efficacy of the irreversible ErbB family blocker Afatinib in epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI)–Pretreated non–small-cell lung cancer patients with brain metastases or leptomeningeal disease, J. Thorac. Oncol., 10: 156–163.
[9]. Westover, D., Zugazagoitia, J., Cho, B. C., Lovly, C. M., Paz-Ares, L. (2018) Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors, Ann. Oncol., 29: i10–i19.
[10]. Kim, Y., Ko, J., Cui, Z., Abolhoda, A., Ahn, J. S., Ou, S. H., Ahn, M. J., Park, K. (2012)The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor, Mol. Cancer Ther., 11: 784–791.
[11]. Liam, K. (2017) Osimertinib as first-line treatment of EGFR mutant advanced nonsmall cell lung cancer, Lung Cancer Res., 6: S62–S66.
[12]. Ward, R. A., Anderton, M. J., Ashton, S., Bethel, P. A., Box, M., Butterworth, S., Colclough, N., Chorley, C. G.,Chuaqui, C., Cross, D. A., Dakin, L. A., Debreczeni, J. E., Eberlein, C., Finlay, M. R. V., Hill, G. B., Grist, M.,Klinowska, T. C., Lane, C., Martin, S., Orme, J. P., Smith, P., Wang, F., Waring, M. J. (2013) Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J. Med. Chem., 56: 7025−7048.
[13]. Abourehab, M. A. S., Alqahtani, A. M., Youssif, B. G. M.,Gouda, A. M. (2021) Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules, 26: 6677
[14]. Cross, D. A. E., Ashton, S.,Ghiorghiu, S., Eberlein, C.,Nebhan, C.,Spitzler, P., Orme, J., Finlay, M. R. V., Ward, R. A., Mellor, M., Hughes, G., Rahi, A., Jacobs, V., Red Brewer, M., Ichihara, E., Sun, J., Jin, H., Ballard, P., Al-Kadhimi, K., Rowlinson, R.,Klinowska, T., Richmond, G.,Cantarini, M., Kim, D.-W., Ranson, M., Pao, W. (2014) AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery, 4: 1046-1061
[15]. Xu, L., Xu, B., Wang, J., Gao, Y., He, X., Xie, T., Ye, X. Y. (2023) Recent advances of novel fourth generation EGFR inhibitors in overcoming C797S mutation of lung cancer therapy, Eur. J. Med. Chem., 245: 114900
[16]. Finlay, M.R.V., Anderton, M., Ashton, S., Ballard, P., Bethel, P.A., Box, M.R., Bradbury, R.H., Brown, S.J., Butterworth, S., Campbell, A. (2014) Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem., 57: 8249–8267.
[17]. Liu, H.,Lv, Y., Li, Y., Cai, J., Chen, J., Qin, Y., Ji, M.(2015)J. Chem. Res., 39: 318-320.
[18]. Zhu, G., Wang, X., Wang, F., Mao, Y., Wang, H. (2017) New and Convergent Synthesis of Osimertinib. J. Heterocycl. Chem., 54: 2898–2901.