







1. Introduction
Since ancient times, humans have been helpless about some traumatic diseases involving bacterial infections. However, humans may have speculated that it is led by some microorganism. Thus, in ancient Egypt would put moldy bread on infected wounds. Natural penicillin is the oldest antibacterial agent derived from fungi, which was first discovered by British scientist Alexander Fleming in 1928, which means Antibiotics were discovered for the first time [1]. Fleming published his findings in 1929. However, the purification from the plate was beyond his capability ten years later, in 1937, while investigating microorganisms and the substances they produced. Howard Florey and Ernst Chain uncovered Fleming’s research and assembled a team of scientists to work solely on the Penicillin Project. After three years of trial and error, they developed a successful but painfully inefficient approach that can produce pure penicillin [1]. For this process, the contaminants or impurities that need to be removed during the purification process of penicillin are culture medium components, such as sugars, amino acids, and other nutrients. Additionally, the metabolic by-products, including organic acids, alcohols, and other secondary metabolites, need to be removed from the penicillin. Residual cell fragments and other microbial materials from the Penicillium mold are present in the culture broth and must be filtered out. Penicillium may produce other compounds besides penicillin, which must be separated during purification. Organic solvents used in extraction and purification steps must be removed to ensure the final product is pure and safe for use.
In 1940, the team led by E. Chain, H. Florey, and N. Heatley of Oxford University solved the stability problem by changing the penicillium culture conditions and optimizing the penicillin extraction process, and finally successfully obtained the pure penicillin product. Subsequently, they used the purified penicillin for animal experiments and obtained the ideal results. In June 1941, Florey decided to take penicillin to the US in the hope of finding a way to scale up production and make the purification of penicillin as efficient as possible. Because America started to attend World War 2, the demand for penicillin rose significantly. America committed to large-scale penicillin production to satisfy the demands of the Armed Forces of the United States, as well as their Allies. On November 19, 1999, the Alexander Fleming Laboratory Museum in London, England, acknowledged the discovery and development of penicillin as a significant milestone in the history of global chemistry. As one of the first and still one of the most widely used antibiotic agents, the discovery of penicillin has significantly contributed to the advancement of medicine and chemistry in subsequent years.
As shown in Figure 1, Penicillin G (penicillin G potassium) is an antibiotic prescribed for the treatment of bacterial infections. It is used for treating pneumonia, strep throat, staph infection, diphtheria, meningitis, gonorrhea, and syphilis. It may be administered to prevent heart valve infection before dental procedures for people with certain cardiac conditions. The mechanism of Penicillin is inhibiting bacterial cell wall synthesis to play a bactericidal effect the structure of penicillin and the components of the cell wall are similar to D-alanine-D-alanine in the mucopepine structure, which can compete with the latter transpeptidase, hinder the formation of mucopeptide, resulting in the defect of the cell wall, make bacteria lose the cell wall penetration barrier, and play a killing effect on bacteria.
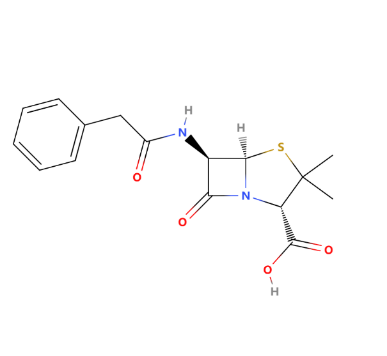
Figure 1: The chemical structure of penicillin G
2. Semisynthetic penicillin
Compared with natural penicillin, semisynthetic penicillin still keeps the 6-aminopenicillins acid, but with the modification of other functional groups, as shown in Figure 2. As for natural penicillin, the main disadvantage is the fragment β-lactam and limited anti-bacterial spectrum. Thus, the researcher attempts to protect the β-lactam from acid-degradation or enzyme-degradation, which could be attributed to the introduction of various R groups (large side chain groups or attracting electron groups). According to the different functionalities of R groups, semisynthetic penicillin can be further divided into five types: acid-resistant, enzyme-resistant, broad-spectrum, anti-pseudomonas aeruginosa, and anti-gram-negative penicillin.
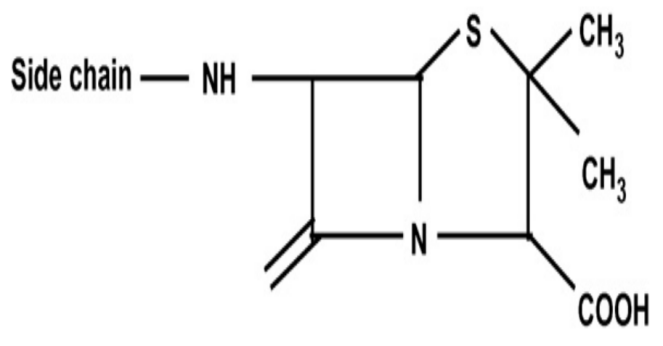
Figure 2: The common chemical structure of semisynthetic penicillin. According to the application, the side chain (R) groups can be various functional groups
2.1. Acid-resistant penicillins
The acid degradation of β-lactam could damage the bioactivity of β-lactam antibiotics. Thus, developing acid-resistant penicillin could be a promising candidate for the next generation of antibiotics. The introduction of large side chain groups or attracting electron groups can help stabilize the β-lactam.
As a typical acid-resistant penicillin, Amoxicillin has been synthesized since 1960 (GlaxoSmithKline plc.). As shown in Figure 3, the R group is an amino acid as an electron-withdrawing group to make the structure more stable. The oral bioavailability of amoxicillin was 70-90%, with peak plasma levels occurring within 1 to 2 hours. Then, the amoxicillin mainly distributes into many tissues, including the liver, lungs, prostate (human), muscle, bile, ascitic, pleural and synovial fluids, and ocular fluids [2].
Amoxicillin has some common side effects, such as skin reactions, which can range from mild rashes to more serious conditions like urticaria (hives) or, rarely, Stevens-Johnson syndrome. Gastrointestinal symptoms often include nausea, vomiting, diarrhea, and abdominal discomfort. The antibiotic can disrupt the normal gut flora, leading to some side effects. For example, hepatic, liver-related side effects are less common but can include elevated liver enzymes and jaundice in some cases. Haematological reactions that have specific manifestations show that Amoxicillin may cause blood-related issues, such as eosinophilia (increased eosinophils), thrombocytopenia (low platelet count), or, very rarely, agranulocytosis (a severe reduction in white blood cells).
Amoxicillin in dilute aqueous solution was found to follow a first-order or pseudo-first-order degradation rate at constant pH with a minimum rate at about pH 6. Amoxicillin degradation was subject to catalysis by phosphate and citrate buffers, with a 10-fold increase in rate with phosphate [3].
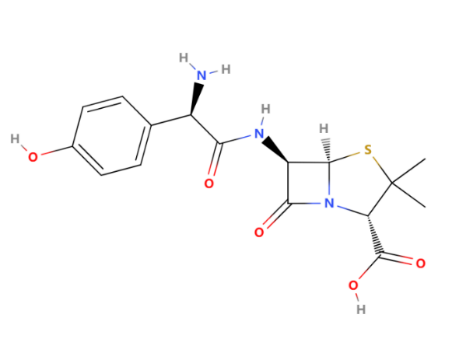
Figure 3: The chemical structure of amoxicillin
2.2. Enzyme-resistant penicillins
Developing enzyme-resistant penicillin primarily aims at addressing the problem of bacterial resistance to antibiotics. Enzymes produced by bacteria, such as β-lactamases, can break down the β-lactam ring of penicillin, rendering the antibiotic ineffective. To overcome this issue, scientists have developed enzyme-resistant penicillin, which can withstand such enzymatic degradation and enhance its therapeutic efficacy. Enzyme-resistant penicillins have the following characteristics. For example, resisting Beta-lactamase means that the penicillins are resistant to degradation by beta-lactamase enzymes that are produced by certain bacteria to inactivate beta-lactam antibiotics. Enzyme resistance allows penicillins to remain effective against beta-lactamase-producing bacteria.
As one of the most widely used Enzyme-resistant penicillins, Nafcillin was developed in the 1960s by researchers at the pharmaceutical company Beecham, now part of Glaxo Smith Kline. As shown in Figure 4, the 2-Ethoxy-1-naphthyl group is its R group, which protects the beta-lactam ring from being attacked by beta-lactamase enzymes. Nafcillin has activity against gram-positive bacteria. Nafcillin is primarily against gram-positive bacteria, especially Staphylococcus aureus. Nafcillin is commonly used to treat infections, such as skin and soft tissue infections, as well as bone infections. Nafcillin exhibits highly effective effects in fighting against methicillin-sensitive Staphylococcus aureus but not on methicillin-resistant Staphylococcus aureus because MRSA has additional resistance mechanisms beyond beta-lactamase production.
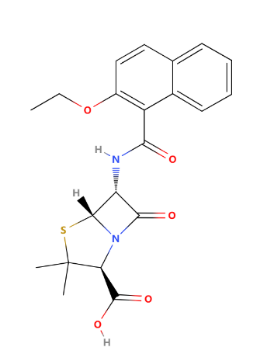
Figure 4: The chemical structure of Nafcillin
2.3. Broad-spectrum penicillins
Due to penicillin G's limited antibiotic effects (mainly fighting against gram-positive bacteria), a broad-spectrum penicillin should be developed to fight both gram-positive and gram-negative bacteria. In this case, broad-spectrum penicillins can be effectively applied in clinical applications, such as patients with multiple bacterial infections.
As one of the most well-known broad-spectrum penicillin, Hetacillin has a feature that Hetacillin itself is not active; instead, it is metabolized into ampicillin (shown in Figure 6) by quickly splits off acetone, which then exerts the antibacterial effect. The half-life of heta cillin at 37 C at pH 7.0 has been estimated at 15 to 30 min. In none of the previous studies could the authors demonstrate an independent antibiotic action of hetacillin, which could not possibly be attributed to ampicillin [4].
The structure of hetacillin is shown in Figure 5. Hetacillin exhibits broad-spectrum activity against a variety of gram-positive (Streptococcus pneumoniae, Streptococcus pyogenes) and some gram-negative bacteria (Haemophilus influenzae). Common side effects may include gastrointestinal disturbances, such as nausea and diarrhoea; allergic reactions, including rash and anaphylaxis; and, in rare cases, hematologic effects like leukopenia and thrombocytopenia. Figure 6 shows the structure of ampicillin. So, compared with ampicillin, hetacillin is the precursor drug of ampicillin, which chemically structurally adds an acetylated 6-hydroxyl group, making it more stable. Hetacillin is developed to have enhanced gastrointestinal stability and bioavailability, overcoming the potential gastrointestinal side effects.
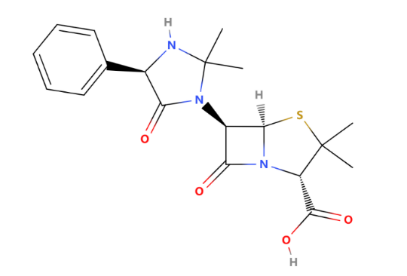
Figure 5: The structure of Hetacillin
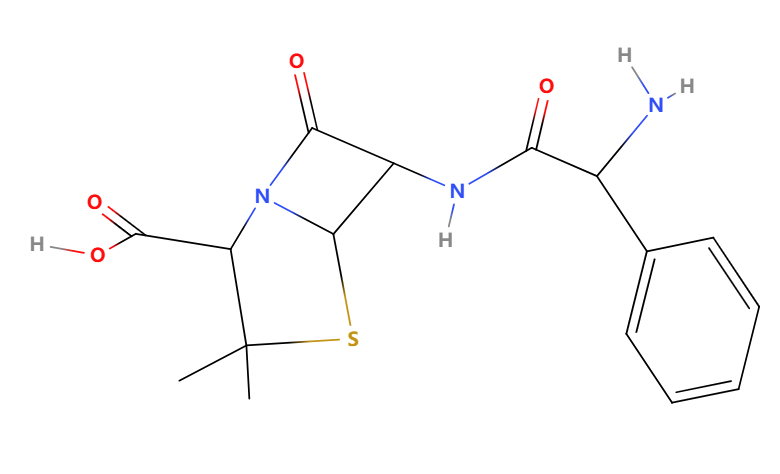
Figure 6: The structure of ampicillin
2.4. Anti-pseudomonas aeruginosa penicillins
Pseudomonas aeruginosa is a common opportunistic pathogen that is notorious for its natural and acquired resistance to many antibiotics. Pseudomonas aeruginosa particularly appears in patients with compromised immune systems, such as those with cancer, organ transplant recipients, and individuals on long-term immunosuppressive therapy. It can cause severe infections like pneumonia, bloodstream infections, urinary tract infections, and wound infections, especially in burn victims. If not treated promptly, these infections can lead to serious complications and even death. The antibacterial spectrum of common penicillin does not include Pseudomonas aeruginosa and the serious acquired resistance of Pseudomonas aeruginosa. The acquired resistance limits the antibiotic effects of standard antibiotics against Pseudomonas aeruginosa. Therefore, it is necessary to develop the anti-Pseudomonas aeruginosa penicillins.
As one of the most potent penicillins against Pseudomonas aeruginosa, Piperacillin is often combined with tazobactam, a beta-lactamase inhibitor, to form piperacillin/tazobactam (Zosyn), which extends its spectrum of activity against beta-lactamase-producing bacteria. Piperacillin is a broad-spectrum, beta-lactam antibiotic that belongs to the ureidopenicillin subclass of penicillins (Figure 7).
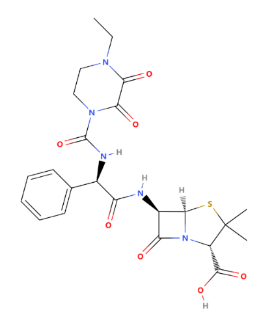
Figure 7: The chemical structure of piperacillin
It is known for its wide range of activity against gram-positive and gram-negative bacteria, including Pseudomonas aeruginosa, a notoriously difficult-to-treat gram-negative pathogen. Piperacillin exhibits a wider spectrum of activity and, in some cases, greater potency against gram-negative organisms than the other members of the penicillin group. Piperacillin at concentrations of 32 mg/L has inhibited around 80% of the most clinically important gram-negative organisms, whilst concentrations of < 128 mg/L are required to inhibit more than 90% of these organisms. Haemophilus influenzae and Neisseria gonorrhoeae have demonstrated marked sensitivity to piperacillin, whilst good activity against Proteus (mirabilis and indole-positive) and Salmonella spp. has been recorded. Pseudomonas aeruginosa has shown greater susceptibility to piperacillin than carbenicillin or ticarcillin with minimal inhibit concentration 90% values of around 32 mg/L, but the susceptibility of the Pseudomonas species was a little lower with MIC90 values of around 64 mg/L. Piperacillin inhibits 90% of most strains of anaerobic bacteria at concentrations of 8 mg/L or less. However, Bacteroides fragilis and Bacteroides species required concentrations within the range of 32 to > 128 mg/L. Activity against B. fragilis varied but was generally slightly greater than that of the other penicillins [5]. However, the side effects caused by Piperacillin cannot be ignored. Gastrointestinal symptoms (nausea, diarrhea), allergic reactions (rash, anaphylaxis), and, occasionally, hematologic effects (thrombocytopenia, neutropenia) may emerge in some people. Although piperacillin has broad-spectrum activity, resistance can occur, particularly in the presence of beta-lactamase-producing organisms. However, combustion with other antibiotics can calm it. Anti-Pseudomonas aeruginosa penicillins are critical in the management of serious infections caused by this notoriously resistant pathogen. Their use is often reserved for severe infections where Pseudomonas aeruginosa is a known or suspected pathogen, and they are frequently used in combination with other antibiotics to enhance efficacy and prevent resistance development. Despite their potency, the emergence of resistance necessitates judicious use and continuous monitoring of antimicrobial susceptibility.
2.5. Anti-gram-negative penicillins
Unlike gram-positive bacteria, Gram-negative bacteria have an outer membrane that makes them more resistant to antibiotics, including natural penicillins. To effectively treat infections caused by gram-negative bacteria, antibiotics need to be developed to penetrate the outer membrane and inhibit the growth of bacteria. Therefore, the development of anti-gram-negative penicillins is necessary. Anti-gram-negative penicillins are a subset of penicillin antibiotics specifically designed to target and treat infections caused by gram-negative bacteria. Due to their great biological activity, anti-gram-negative penicillins are widely used in clinical treatment, especially in hospital-acquired infections, such as pneumonia, urinary tract infections, and bacteremia, especially in patients with weakened immune systems. In addition, there is another thing to notice: the major anti-gram-negative penicillins have large molecular function groups and poor oral absorption. Therefore, most anti-gram-negative bacteria penicillins need to be administered intravenously to ensure effective drug concentrations.
As one of the most widely used anti-gram-negative penicillins, Ticarcillin's structure is shown in Figure 8. Ticarcillin is a beta-lactam antibiotic belonging to the penicillin class. It is a semi-synthetic penicillin derived from 6-aminopenicillanic acid, originally developed by Beecham Pharmaceuticals in the early 1970s. Ticarcillin is effective against a wide range of gram-negative bacteria, including Pseudomonas aeruginosa, Enterobacter, Escherichia coli, and Proteus species. Ticarcillin is often combined with clavulanate (Timentin) to combat beta-lactamase-producing bacteria. Ticarcillin also has great antibacterial activity. The antibacterial activity is similar to that of carbenicillin, with the principal difference being that ticarcillin is consistently 2 to 4 times more active in vitro against Pseudomonas aeruginosa and more active in vivo against experimental Pseudomonas infection. At a concentration of 50μg/ml, ticarcillin is active against about 75% of E. coli and Enterobacter species, 80% of P. aeruginosa and 85% of P. mirabilis and indole-positive Proteus. It has little activity against K. pneumoniae, an organism that is often reported in nosocomial infections. Concentrations of about 16μg/ml are inhibitory to most gram-positive species and for about 25 % of S. faecalis [6].
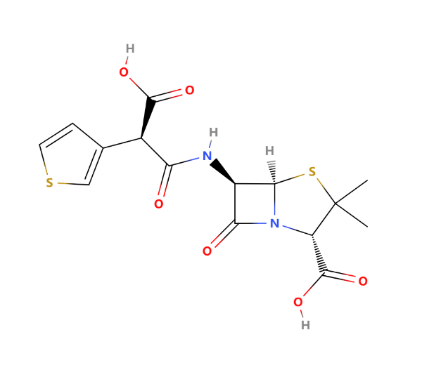
Figure 8: The chemical structure of ticarcillin
Like other penicillins, ticarcillin is bactericidal against susceptible bacteria, the minimum bactericidal concentration generally being 2-fold greater than the inhibitory concentration. About 75% of strains of Bacteroides fragilis are inhibited in vitro by 32 to 64μg/ml of ticarcillin. Lower concentrations of the drug inhibit most other anaerobes. The influence of inoculum size on the susceptibility of bacteria to ticarcillin is similar to that with many other penicillins, although less than with some other antipseudomonal penicillins, and varies according to the bacterial species and strain. Ticarcillin is susceptible to β-lactamase produced by S. aureus and common gram-negative organisms [6]. Thus, Ticarcillin is widely used in the clinical treatment of different microbes, such as lower respiratory tract infections, urinary tract infections, intra-abdominal infections, septicemia, and so on. Although ticarcillin has a broad spectrum of activity, hypersensitivity reactions, gastrointestinal disturbances, possible alterations in blood cell counts, and other side effects need to be noticed even if it is less common. Ticarcillin has been well tolerated by the majority of patients, the most frequently reported adverse effects being skin rash, hypokalaemia, eosinophilia, and pain after intramuscular or phlebitis with intravenous injection. Although dosages of ticarcillin required to treat serious infection may cause platelet dysfunction, there have been few reports of bleeding. Although ticarcillin injection contains 5mEq of sodium per gram, fluid overload has seldom been noted, and ototoxicity and nephrotoxicity have seldom occurred, and then only when ticarcillin was given in combination with an aminoglycoside. There is no evidence that ticarcillin contributes to the nephrotoxicity or ototoxicity reported with combination regimens [6].
3. Cephalosporins
The story of cephalosporins began in 1945 when Italian scientist Giuseppe Brotzu discovered a fungus called Cephalosporium acremonium (later reclassified as Acremonium chrysogenum) off the coast of Sardinia, Italy. Brotzu observed that this fungus produced a substance that was effective against certain gram-positive and gram-negative bacteria, including Salmonella typhi, the causative agent of typhoid fever. Cephalosporin contains a 6-membered dihydrothiazine ring. Substitutions at position 3 generally affect pharmacology; substitutions at position 7 affect antibacterial activity, but these cases are not always true [7].
The key functional groups in cephalosporins include the β-Lactam Ring, which is essential for antibacterial activity, the dihydrothiazine ring which can distinguish cephalosporins from other β-lactam antibiotics like penicillins, acyl side chain, which can improve the antibiotic's spectrum of activity, the most decisive group is R1 and R2 side chains which can not only improve the antibacterial activity but also modifying pharmacokinetic and pharmacodynamic properties. Cephalosporins work by inhibiting bacterial cell wall synthesis. Cephalosporins bind to penicillin-binding proteins in the bacterial cell wall, preventing the cross-linking of peptidoglycan chains, which is essential for cell wall strength and rigidity. This inhibition leads to bacterial cell lysis and death.
3.1. First generation of cephalosporin
The first commercially available cephalosporin, cephalothin (also known as cefalotin), was developed and introduced in the early 1960s. It was the first of the first-generation cephalosporins and was marketed by Eli Lilly and Company. About the spectrum of activity, the first generation of cephalosporin has good activity against gram-positive bacteria such as Streptococcus and Staphylococcus species but limited activity against gram-negative bacteria, primarily effective against Escherichia coli, Klebsiella pneumoniae, and Proteus mirabilis.
Cefazolin
As one of the typical first generations of cephalosporin, Cefazolin is often used for its broad-spectrum activity against gram-positive bacteria and some gram-negative bacteria. The structure of Cefazolin is shown in Figure 9. In particular, it can be used to treat cellulitis, urinary tract infections, pneumonia, infective endocarditis, septic arthritis, and acute cholangitis. It is also used before and after childbirth and before surgery to prevent group B streptococcal infections [8]. The drug is usually administrated intramuscularly or intravenously. To talk about the R group, in cefazolin, the R group is 7-(2-(1H-tetrazol-1-yl) acetamido), which can improve the spectrum of activity and pharmacokinetics. The tetrazole ring is a five-membered ring containing four nitrogen atoms and one carbon atom. This ring contributes to the stability of the cefazolin molecule and its resistance to enzymatic degradation by β-lactamases. The acetamido group (-NH-CO-CH2-) is attached to the tetrazole ring. This group helps form hydrogen bonds with bacterial penicillin-binding proteins, enhancing the antibiotic's binding affinity and effectiveness. Cefazolin may commonly cause side effects such as gastrointestinal discomfort, allergic reactions, and local reactions at the injection site. Although rare, severe side effects can include anaphylaxis and Clostridioides difficile-associated diarrhea. Patients with a known allergy to cephalosporins or penicillins may be at risk for allergic reactions to cefazolin.
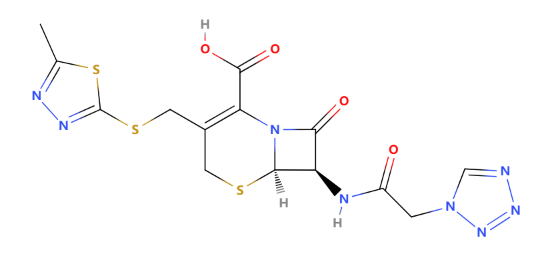
Figure 9: The chemical structure of cefazolin
3.2. Second generation of cephalosporin
Second-generation cephalosporins were developed and introduced primarily in the 1970s and 1980s. Second-generation cephalosporins are a class of antibiotics that belong to the larger group of cephalosporins, which are β-lactam antibiotics. They are used to treat a variety of bacterial infections and are known for their enhanced activity against certain gram-negative bacteria compared to first-generation cephalosporins. Second-generation cephalosporins act on both gram-positive and gram-negative bacteria. However, they are relatively less effective against gram-positive bacteria.
Cefoxitin(shown in Figure 10) is a second-generation cephamycin antibiotic developed by Merck & Co., Inc. from Cephamycin C in the year following its discovery in 1972. It was synthesized to create an antibiotic with a broader spectrum [9]. It is often grouped with the second-generation cephalosporins. Cefoxitin requires a prescription and, as of 2010, is sold under the brand name Mefoxin by Bioniche Pharma, LLC. The generic version of cefoxitin is known as cefoxitin sodium [10]. To talk about the R group contains a β-lactam ring and is resistant to β-lactamase enzymes, which are produced by certain bacteria to inactivate antibiotics. The R group in cefoxitin refers to the side chain attached to the β-lactam ring. For cefoxitin, the specific R group is a 7-alpha-methoxy group and a 3-carbamoyloxy group.
The 7-alpha-methoxy group provides resistance to β-lactamase enzymes, making cefoxitin more stable in the presence of these enzymes compared to other cephalosporins. The 3-carbamoyloxy group contributes to its antibacterial activity and spectrum. This R group configuration gives cefoxitin its unique pharmacological properties and spectrum of activity against bacteria. However, the side effects could not be avoided. Common side effects may include gastrointestinal disturbances, hypersensitivity reactions, and alterations in blood clotting (especially with agents like cefotetan and cefoxitin, which can inhibit vitamin K-dependent clotting factors).
Figure 10: The structure of cephalosporin
3.3. Third generation of cephalosporin
Third-generation cephalosporins are medications used in the management and treatment of gram-negative and gram-positive organisms. They are encompassed among the beta-lactam class of drugs. To talk about the spectrum of Activity, they have a broad spectrum of activity, with enhanced efficacy against gram-negative bacteria. Some agents(e.g., ceftazidime) are effective against Pseudomonas aeruginosa. Good central nervous system penetration (e.g., ceftriaxone, cefotaxime).
The push for the development of third-generation cephalosporins came from the growing need for antibiotics that could effectively target a wider range of gram-negative pathogens, especially those producing beta-lactamases—enzymes capable of breaking down beta-lactam antibiotics and rendering them ineffective. Third-generation cephalosporins were synthesized with modifications to the chemical structure of the cephalosporin nucleus, which enhanced their stability against beta-lactamases and broadened their spectrum of activity. It is one of the most famous third-generation of cephalosporins. The structure is shown in Figure 11.
In this case, the R group (attached to the core beta-lactam structure at the 7-position) is a methoxyimino group, specifically a "CH3OCH═N" group. This group is critical in determining the antibiotic's spectrum of activity and resistance to beta-lactamase enzymes produced by bacteria. Cefotaxime is an antibiotic used to treat several bacterial infections in humans, other animals, and plant tissue cultures [11]. Specifically in humans, it is used to treat joint infections, pelvic inflammatory disease, meningitis, pneumonia, urinary tract infections, sepsis, gonorrhea, and cellulitis.[11] It is given either by injection into a vein or muscle. About the antibacterial activity, Cefotaxime exhibits both a wider spectrum and greater activity against gram-negative aerobic bacteria than 'first generation' or 'second generation' cephalosporins, is generally more active than cefoperazone except against Pseudomonas aeruginosa, and similar in activity to moxalactam. A multicentre study in the USA found that over 91% of 6000 clinical isolates of Enterobacteriaceae were inhibited by 0.5 μg/ml or less cefotaxime. This antibiotic is active against many cephalothin-resistant and gentamicin-resistant Enterobacteriaceae and against some strains that show multiple drug resistance. Cefotaxime is also active at very low concentrations (MIC90 ⩽ 0.06 μg/ml) against β-lactamase-producing and non-producing strains of Haemophilus influenzae and Neisseria gonorrhoeae. Although cefotaxime tends to be less active than cefoxitin against Bacteroides fragilis, it inhibits most other anaerobic bacteria at low concentrations [12].
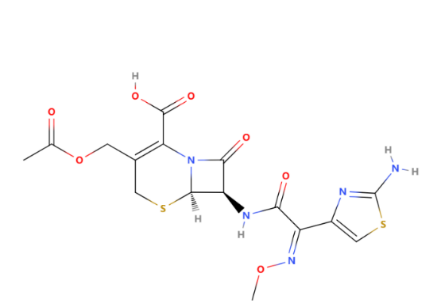
Figure 11: The structure of cefotaxime
3.4. Fourth generation of cephalosporin
Fourth-generation cephalosporins are a group of antibiotics structurally related to third-generation cephalosporins but possess an extra ammonium group1. The only available fourth-generation cephalosporin is cefepime (Maxipime), which is active against various gram-positive and gram-negative bacteria, including Pseudomonas aeruginosa, Enterobacter, and Citrobacte. The fourth-generation cephalosporins- ins (represented by cefepime, cefoselis, cefclidin, cefozopran, and cefquinome) have broad antibacterial spectra that encompass gram-negative bacteria, including P. aeruginosa and gram-positive bacteria. Their enhanced activities against gram-negative bacteria can be attributed to their poor affinity for and increased stability to the Bush group 1 P-lactamases and their more rapid penetration across the outer membrane of bacterial cells. Collectively, the third- and fourth-generation cephalosporins have been referred to as extended-spectrum cephalosporins. Limited microbiological and clinical data on cefoselis, cefclidine, cefquinome, and cefozopran are published in the English literature, and data on these compounds are included in this review when available. Cefepime and cefpirome are used clinically in several countries for the treatment of moderate to severe bacterial infections. However, cefepime is currently the only fourth-generation cephalosporin approved for clinical use in the United States [13]. So, when we refer to the fourth generation of cephalosporin, it always refers to cefepime. Its structure is shown in Figure 12
Cefepime is a fourth-generation cephalosporin antibiotic. Cefepime has an extended spectrum of activity against gram-positive and gram-negative bacteria, with greater activity against both types of organisms than third-generation agents. A 2007 meta-analysis suggested that when data from trials were combined, mortality was increased in people treated with cefepime compared with other β-lactam antibiotics [14]. In response, the U.S. Food and Drug Administration performed its meta-analysis, which found no mortality difference.
About the R group,the R1 group at the 7-position of cefepime is the "2-[2-[(aminocarbonyl)oxy]imino]-2-(pyrrolidinium-1-yl)acetamido" group, while the R2 group at the 3-position is "1-methyl-1-pyrrolidinium." These groups are crucial for cefepime's pharmacological properties, including its antibacterial activity and resistance to beta-lactamase enzymes [15].
Cefepime is usually administered intravenously or intramuscularly. It is well-distributed throughout the body, including in the cerebrospinal fluid, which makes it useful for treating central nervous system infections. Moreover, it can be used to treat pneumonia, urinary tract infections, skin infections, intra-abdominal infections, febrile neutropenia, and so on. It is not hard to think that, like all medications, cefepime can cause side effects, including allergic reactions, gastrointestinal upset, and, in rare cases, neurological effects such as seizures, even thrombocytopaenia, particularly in patients with renal impairment [16].
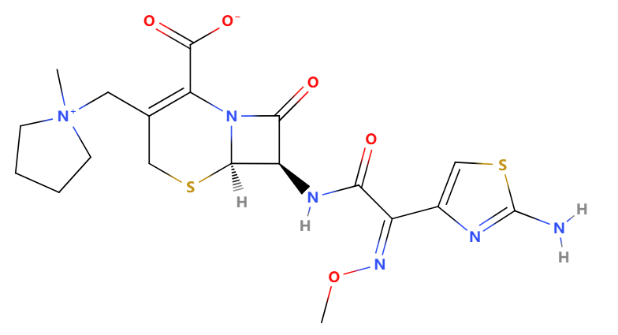
Figure 12: The structure of cefepime
4. Other β lactam antibiotics
Other β lactam antibiotics, all containing the β lactam ring as the main functional group, differ from the positions of the R1, R2, and R3 variable functional groups, the five-membered/six-membered ring, and the S and O atoms.
4.1. Oxycephems
Oxycephems are a kind of β-lactam antibiotics similar to cephalosporins. They are characterized by a unique structural feature: an O atom replacing an S atom in the cephem nucleus. This modification can influence the antibiotic's spectrum of activity, stability, and resistance to β-lactamases, as shown in Figure 13.
Cefmetazole is a cephamycin antibiotic, usually grouped with the second-generation cephalosporins. It is used to treat a variety of bacterial infections by interfering with the synthesis of the bacterial cell wall, leading to cell lysis and death. This antibiotic is particularly effective against a range of gram-positive and gram-negative bacteria, including those that produce beta-lactamase enzymes, which often confer resistance to other beta-lactam antibiotics.
Cefmetazole was introduced into clinical use in the late 20th century. The exact year of its development and approval can vary by country, but it was generally introduced in the 1970s and 1980s. Cefmetazole belongs to the broader class of cephalosporins, Fujisawa Pharmaceutical Co., Ltd., a Japanese pharmaceutical company initially discovered in the fungus Cephalosporium acremonium. This discovery led to the development of a range of cephalosporin antibiotics.
Like other cephalosporins, cefmetazole contains a beta-lactam ring crucial for antibacterial activity. However, it has a unique side chain at the 3-position of the cephem nucleus, which includes a methoxy group and a thiazole ring. This structural modification enhances its stability against beta-lactamase enzymes produced by certain bacteria, which can inactivate other beta-lactam antibiotics. β-Lactamases failed to hydrolyze cefmetazole significantly, and cefmetazole is considered the most stable of the cephamycin drugs. Some Bacteroides strains produce β-lactamases that are uniquely inhibited by cefmetazole [17].
In the process of research and development, It was found effective against a wide range of gram-positive and gram-negative bacteria, including some beta-lactamase-producing organisms. It is especially useful in treating infections where second-generation cephalosporins are indicated, such as respiratory, urinary, and intra-abdominal infections. Like other antibiotics, cefmetazole can cause a range of side effects like Nausea, Vomiting, Diarrhea, and abdominal pain. Especially cefmetazole can trigger hypoprothrombinemia, but hemolytic anemia is not a side effect frequently that occurs [18]. Additionally, prolonged use of cefmetazole, as with other antibiotics, may result in the overgrowth of resistant organisms.
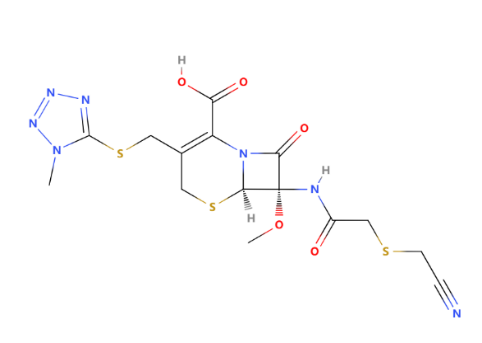
Figure 13: The structure of cefmetazole
4.2. Carbapenems
The carbapenem class of antibiotics was derived from thienamycin, a naturally occurring compound produced by the bacterium Streptomyces cattleya. Thienamycin was first isolated in 1976 and was noted for its potent antibacterial activity and stability against beta-lactamases. Scientists at Merck & Co., an American pharmaceutical company, conducted the discovery and initial research on thienamycin and its derivatives.
Thienamycin was found to be chemically unstable, leading researchers to develop more stable derivatives. Imipenem was the first carbapenem developed for clinical use. It was synthesized by adding an N-formimidoyl group to thienamycin and introduced into clinical practice in the 1980s. Other carbapenems, such as meropenem, ertapenem, and doripenem, followed, each with unique pharmacokinetic and pharmacodynamic properties.
Carbapenems are effective against a wide range of gram-positive bacteria, including Streptococcus species, Staphylococcus aureus (including some MRSA strains), and Enterococcus faecalis. They have broad activity against many gram-negative bacteria, including Escherichia coli, Klebsiella species, Proteus species, and Pseudomonas aeruginosa. Even effective against many anaerobic bacteria, including Bacteroides fragilis and Clostridium species. So, the advantage of carbapenems is in evidence. First, Broad Spectrum could be evident by being effective against a wide range of pathogens, including multi-drug-resistant organisms. Second, they have Beta-Lactamase stability resistant to many beta-lactamases that typically degrade other beta-lactam antibiotics. Moreover, potent activity is often reserved for severe infections due to their potency and broad spectrum.
However, the development of carbapenemase also faces some challenges. Carbapenemase Production: Enzymes such as KPC (Klebsiella pneumoniae carbapenemase), NDM (New Delhi metallo-beta-lactamase), and OXA-48 can hydrolyze carbapenems, rendering them ineffective.Efflux Pumps: Some bacteria possess efflux systems that can pump carbapenems out of the cell.Porin Channel Alterations: Changes in the bacterial cell membrane can prevent carbapenems from entering the bacterial cell.
Imipenem is the active antibiotic, while cilastatin is a renal dehydropeptidase inhibitor that prevents the breakdown of imipenem in the kidneys (Figure 14). Used for severe infections, including intra-abdominal infections, pneumonia, and sepsis. For imipenem, the key side chain, which can be considered the R group, is the N-formimidoyl group. This specific group differentiates imipenem from other carbapenems and plays a role in its biological activity and pharmacokinetic properties. The pharmacologic properties of imipenem are similar to those of third-generation cephalosporins, which are cleared by the kidney and are not bound extensively to serum proteins. The extent of serum protein binding for imipenem is 20%. In healthy volunteers with normal renal function, the serum half-life is approximately 1 hour. In adults of average size with normal renal function, a 1,000-mg dose administered intravenously during a 30-minute period produces peak levels of 55 to 65 μg/ml.
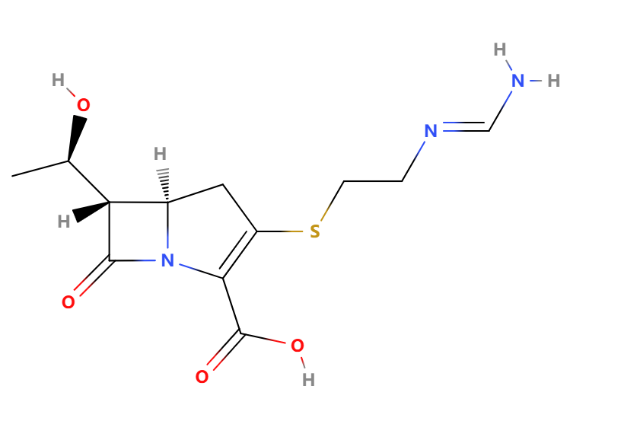
Figure 14: Imipenem
4.3. Monocyclic β-lactams
Monocyclic β-lactams are a distinctive group within the broader category of β-lactam antibiotics. Unlike other β-lactams, monobactams have a unique structure that provides stability against certain β-lactamases. They specifically target gram-negative bacteria and evade the β-lactamase enzymes that hydrolyze the β-lactam ring.
The most widely used monocyclic β-lactam is aztreonam (Figure 15), developed in the 1980s. Aztreonam was the only monobactam available on the market. It has a narrow spectrum of activity, mainly against gram-negative aerobic bacteria, making it particularly useful in treating infections caused by these organisms. Aztreonam's structure allows it to bind specifically to the penicillin-binding proteins of gram-negative bacteria, inhibiting cell wall synthesis and leading to bacterial death. Although the spectrum of activity is narrow, the speciality and high efficiency make aztreonam can be used to treat infections by susceptible bacteria without disrupting the patient's microbiota [20].
Clinical experience with aztreonam is extensive, and urinary tract infections represent the most frequent types of infection that are treated with this agent. 50-52 Pooled data from studies conducted around the world reveal clinical response rates to aztreonam monotherapy for urinary tract infections above 80 per cent [21,22]
Because oral utilization is only about one percent which means aztreonam essentially is not absorbed from the gastrointestinal tract [23], aztreonam is given by intravenous or intramuscular injection or by inhalation. It is rapidly and completely absorbed following intramuscular. A 500 mg IM aztreonam dose produces 21 to 27 f serum concentrations. Lg per ml at 1 hour, 3.8 to 5.9 f.Lg per ml at 6 hours, 1.5 to 3.3 f.Lg per ml at 8 hours, and 0.1 to 1. 7 f.Lg per ml at 12 hours. A 1 g IM dose yields peaks of 3.5 f. Lg per ml at 8 hours and 0.7 f.Lg per ml at 12 hours [24 ].
Aztreonam is generally well-tolerated, with a safety profile that makes it a good option for patients with allergies to other β-lactams, such as penicillins and cephalosporins. However, some will have side effects, such as Injection site reactions, Gastrointestinal disturbances (nausea, vomiting, diarrhea), Rash, and Liver enzyme elevation.
The future of β-lactam antibiotics will prioritize innovation in antimicrobial resistance, encompassing the development of new antibiotics, enhancement of β-lactamase inhibitors, promotion of antimicrobial combination therapy, implementation of precision antibiotic therapy, exploration of new routes of administration, and reinforcement of drug resistance monitoring and management. These endeavours are focused on enhancing treatment results, delaying the emergence of drug resistance, and guaranteeing the ongoing efficacy of antibiotics in medical practice.
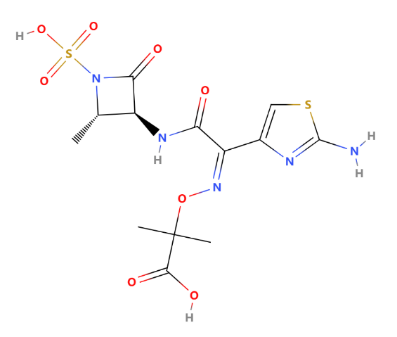
Figure 15: Structure of aztreonam
5. Conclusion
Overall, the presence of the β-lactam ring is crucial for the efficacy of β-lactam antibiotics. However, the β-lactam ring is unstable and may undergo acid hydrolysis or enzymatic degradation, leading to the inactivation of β-lactam antibiotics and even developing antibiotic resistance. Therefore, researchers have attempted to stabilize the structure of β-lactam antibiotics by modifying the R groups near the β-lactam ring, making it less susceptible to acid and enzymatic degradation. Modifying the R group can also broaden the antibacterial spectrum of β-lactam antibiotics. These R groups have electron-withdrawing effects and provide significant steric hindrance. Based on the differences in the R groups, β-lactam antibiotics can be classified into penicillins, cephalosporins, carbapenems, and monobactams.
References
[1]. Fleming, A. (1929). On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzae. British Journal of Experimental Pathology, 10(3), 226-236
[2]. Elizalde-Velázquez, A., Gómez-Oliván, L. M., Galar-Martínez, M., Islas-Flores, H., Dublán-García, O., & SanJuan-Reyes, N. (2016). Amoxicillin in the Aquatic Environment, Its Fate and Environmental Risk. In InTech eBooks.
[3]. KAUR, S. P., RAO, R., & NANDA, S. (2011). AMOXICILLIN: A BROAD SPECTRUM ANTIBIOTIC. In International Journal of Pharmacy and Pharmaceutical Sciences (Vols. 3–3, pp. 30–37)
[4]. Faine S, Harper M. 1973. Independent Antibiotic Actions of Hetacillin and Ampicillin Revealed by Fast Methods. Antimicrob Agents Chemother 3:.
[5]. Holmes, B., Richards, D.M., Brogden, R.N. et al. Piperacillin. Drugs 28, 375–425 (1984)
[6]. Brogden, R.N., Heel, R.C., Speight, T.M. et al. Ticarcillin: A Review of its Pharmacological Properties and Therapeutic Efficacy. Drugs 20, 325–352 (1980).
[7]. Prince, A. (n.d.). CEPHALOSPORINS AND VANCOMYCIN.
[8]. The American Society of Health-System Pharmacists. [2016-12-08]
[9]. Gootz, T. (1990). Discovery and Development of New Antimicrobial Agents. In Clinical Microbiology Reviews (Vol. 3, pp. 13–31). American Society for Microbiology.
[10]. Supplement Approval for Mefoxin (Cefoxitin for Injection, USP). (2011). In Food and Drug Administration
[11]. Supplement Approval for Mefoxin (Cefoxitin for Injection, USP). (2011). In Food and Drug Administration
[12]. Carmine, A.A., Brogden, R.N., Heel, R.C. et al. Cefotaxime. Drugs 25, 223–289 (1983).
[13]. Joan C. Fung-Tome, Ph.D. Department of Microbiology Bristol-Myers Squibb Company Wallingford, CT 06492.
[14]. Yahav D, Paul M, Fraser A, Sarid N, Leibovici L (May 2007). "Efficacy and safety of cefepime: a systematic review and meta-analysis". The Lancet. Infectious Diseases. 7 (5): 338–348.doi:10.1016/S1473-3099(07)70109-3.
[15]. Endimiani, A., Perez, F., & Bonomo, R. A. (2008). Cefepime: a reappraisal in an era of increasing antimicrobial resistance. In Expert Rev Anti Infect Ther (Vols. 6–6, pp. 805–824).
[16]. Endimiani, A., Perez, F., & Bonomo, R. A. (2008). Cefepime: a reappraisal in an era of increasing antimicrobial resistance. In Expert Rev Anti Infect Ther (Vols. 6–6, pp. 805–824).
[17]. Ronald N. Jones, Review of the in-vitro spectrum and characteristics of cefmetazole (CS-1170), Journal of Antimicrobial Chemotherapy, Volume 23, Issue suppl_D, 1989, Pages 1–12.
[18]. Fukuda M, Nabeta M, Oya S, et al. Severe drug-induced immune hemolytic anemia due to cefmetazole: A case report[J]. International Journal of Clinical Pharmacology and Therapeutics, 2022, 60(1): 52.
[19]. HELLINGER, W. C., & BREWER, N. S. (1991, October). Imipenem. In Mayo Clinic Proceedings (Vol. 66, No. 10, pp. 1074-1081). Elsevier.
[20]. HELLINGER, W. C., & BREWER, N. S. (1991, October). Imipenem. In Mayo Clinic Proceedings (Vol. 66, No. 10, pp. 1074-1081). Elsevier.
[21]. Madsen PO, Nielsen KT, and Craversen PH: Aztreonam: a critical evaluation of the first monobactam antibiotic in the treatment of urinary tract infections. 1 Urol 140: 925 0988).
[22]. Stutman HR: Clinical experience with aztreonam, Pediatr Infect Dis J (Suppl 9) 8: S109 (1989).
[23]. Swabb EA, Sugerman AA, Stern M: Oral bioavailability of the monobactam aztreonam (SQ 26,776) in healthy subjects. Antimicrob Agents Chemother 2:3:.548-.5,50, 1983
[24]. Swabb EA: Review of the clinical pharmacology of the monobactam antibiotic aztreonam. Am J Med 78(Suppl 2A):11-11), 1911,5
Cite this article
Zhang,Z. (2025). The History, Present, and Future of β-lactam Antibiotics. Theoretical and Natural Science,116,37-51.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 3rd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Fleming, A. (1929). On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzae. British Journal of Experimental Pathology, 10(3), 226-236
[2]. Elizalde-Velázquez, A., Gómez-Oliván, L. M., Galar-Martínez, M., Islas-Flores, H., Dublán-García, O., & SanJuan-Reyes, N. (2016). Amoxicillin in the Aquatic Environment, Its Fate and Environmental Risk. In InTech eBooks.
[3]. KAUR, S. P., RAO, R., & NANDA, S. (2011). AMOXICILLIN: A BROAD SPECTRUM ANTIBIOTIC. In International Journal of Pharmacy and Pharmaceutical Sciences (Vols. 3–3, pp. 30–37)
[4]. Faine S, Harper M. 1973. Independent Antibiotic Actions of Hetacillin and Ampicillin Revealed by Fast Methods. Antimicrob Agents Chemother 3:.
[5]. Holmes, B., Richards, D.M., Brogden, R.N. et al. Piperacillin. Drugs 28, 375–425 (1984)
[6]. Brogden, R.N., Heel, R.C., Speight, T.M. et al. Ticarcillin: A Review of its Pharmacological Properties and Therapeutic Efficacy. Drugs 20, 325–352 (1980).
[7]. Prince, A. (n.d.). CEPHALOSPORINS AND VANCOMYCIN.
[8]. The American Society of Health-System Pharmacists. [2016-12-08]
[9]. Gootz, T. (1990). Discovery and Development of New Antimicrobial Agents. In Clinical Microbiology Reviews (Vol. 3, pp. 13–31). American Society for Microbiology.
[10]. Supplement Approval for Mefoxin (Cefoxitin for Injection, USP). (2011). In Food and Drug Administration
[11]. Supplement Approval for Mefoxin (Cefoxitin for Injection, USP). (2011). In Food and Drug Administration
[12]. Carmine, A.A., Brogden, R.N., Heel, R.C. et al. Cefotaxime. Drugs 25, 223–289 (1983).
[13]. Joan C. Fung-Tome, Ph.D. Department of Microbiology Bristol-Myers Squibb Company Wallingford, CT 06492.
[14]. Yahav D, Paul M, Fraser A, Sarid N, Leibovici L (May 2007). "Efficacy and safety of cefepime: a systematic review and meta-analysis". The Lancet. Infectious Diseases. 7 (5): 338–348.doi:10.1016/S1473-3099(07)70109-3.
[15]. Endimiani, A., Perez, F., & Bonomo, R. A. (2008). Cefepime: a reappraisal in an era of increasing antimicrobial resistance. In Expert Rev Anti Infect Ther (Vols. 6–6, pp. 805–824).
[16]. Endimiani, A., Perez, F., & Bonomo, R. A. (2008). Cefepime: a reappraisal in an era of increasing antimicrobial resistance. In Expert Rev Anti Infect Ther (Vols. 6–6, pp. 805–824).
[17]. Ronald N. Jones, Review of the in-vitro spectrum and characteristics of cefmetazole (CS-1170), Journal of Antimicrobial Chemotherapy, Volume 23, Issue suppl_D, 1989, Pages 1–12.
[18]. Fukuda M, Nabeta M, Oya S, et al. Severe drug-induced immune hemolytic anemia due to cefmetazole: A case report[J]. International Journal of Clinical Pharmacology and Therapeutics, 2022, 60(1): 52.
[19]. HELLINGER, W. C., & BREWER, N. S. (1991, October). Imipenem. In Mayo Clinic Proceedings (Vol. 66, No. 10, pp. 1074-1081). Elsevier.
[20]. HELLINGER, W. C., & BREWER, N. S. (1991, October). Imipenem. In Mayo Clinic Proceedings (Vol. 66, No. 10, pp. 1074-1081). Elsevier.
[21]. Madsen PO, Nielsen KT, and Craversen PH: Aztreonam: a critical evaluation of the first monobactam antibiotic in the treatment of urinary tract infections. 1 Urol 140: 925 0988).
[22]. Stutman HR: Clinical experience with aztreonam, Pediatr Infect Dis J (Suppl 9) 8: S109 (1989).
[23]. Swabb EA, Sugerman AA, Stern M: Oral bioavailability of the monobactam aztreonam (SQ 26,776) in healthy subjects. Antimicrob Agents Chemother 2:3:.548-.5,50, 1983
[24]. Swabb EA: Review of the clinical pharmacology of the monobactam antibiotic aztreonam. Am J Med 78(Suppl 2A):11-11), 1911,5