







1. Introduction
Drug targets are mainly applied to interfere with the action of disease-causing genes, and the development of DNA sequencing technology has enabled rapid screening of disease-causing genes, so that early intervention is possible for certain genetic diseases. However, most hereditary diseases do not rely on a single disease-causing gene to develop, but rather multiple genes acting together. The risk of developing the disease is not 100%, but can be calculated based on the location of the parents' disease-causing gene. For example, mutations in families containing the BRCA gene increase the risk of cancer, but not everyone with a mutated BRCA gene will develop cancer. In addition to the disease-causing gene, the environment is also an influencing factor for the disease, such as prolonged exposure to radiation or highly toxic environments, which can cause normal genes to mutate into disease-causing genes, so drug targets may also include molecular pathways related to the environment. Mutations in certain alleles can also lead to an increased risk of disease, and these require targeted medicine for treatment.
Drug discovery and development can broadly follow two different paradigms —physiology-based drug discovery and target-based discovery. The difference between them is the point in time when the drug target is identified [1]. The former is more focused on following physiological readouts in animal or cellular experiments, then screening and analyzing the lead compounds based on the data, while the identification of drug targets is later inferred based on the pharmacological properties of the lead compounds [2]. The latter studies from the way of gene, identifying target functions and exploring their role in a specific disease. This process is complex because of the genetic diversity. The development of genomics allows these two approaches to sometimes work in tandem. Currently, all available therapies combined hit only about 400 different drug targets [3], and have great potential for future development.
Since the first generation of targeted drugs came into use, targeted therapy has occupied an important place in drug history by virtue of their high efficacy and low side effects, RNAi drugs are a good example. The wide range of audience makes the safety and other aspects of targeted drugs particularly important. Whether the disease target is effective in humans, how to find the hits and screen the leads are questions that should be considered by those involved. Although most disease targets discovered will be screened out at the validation and screening stage, pharmacologists are still fascinated by the great role that these targets play in treating diseases. The importance of drug target validation is to demonstrate that a molecular target plays a key role in the disease pathway and the validated target needs to have some therapeutic effect. Validation of molecular target effectiveness in vitro usually takes precedence over molecular therapeutic validation in vivo, and the data from both validations together define the clinical potential of the target. Target validation also includes studies on animals or based on disease-related cellular models. These models can provide information about how an organism responds to a drug after it is taken or injected and can test important information about the drug in the body, including drug ADME (absorption distribution metabolism and excretion). Thus, it helps to predict what is likely to happen to patients after taking drugs. There are many types of viable disease targets, including: receptors, proteins and enzymes, DNA, RNA and ribosomal targets. One of the more novel, and one that many pharmaceutical companies are focusing on, is the RNA target. This paper describes how to discover and assess disease targets and analyses the mechanism of RNAi technology in treating rare diseases and its delivery system. Meanwhile, the article analyzes the advantages and shortcomings of RNAi technology, and provides an outlook on the future of RNAi technology.
2. Results
The process of taking a drug from validation of target effectiveness to application is described in details below.
2.1. Validation
Gene knock-out/in——The establishment of transgenic models can be achieved by target gene knock-out/in for adult animals, which is an important experimental validation method. The disease model is one of the transgenic models that can be combined with gene knock-out/in. The knock-out of target genes facilitates the study of the effects of regulatory drug targets. Knock-out/in will activate gene expression of target genes, sometimes improving or even reversing a sign of disease, so knockouts are also used to create disease models. However, gene knock-out/in can sometimes be fatal for organisms. A protein is said to be essential for an organism if a knock-out or mutation results in lethality or infertility, for instance, switching on or restoring the function of cell cycle genes in postmitotic cells often leads to cell death [4]. This would also be a good target validation strategy if gene function could be modified by knock-out/in and then restored in a specific organ or tissue.
RNAi and antisense DNA/RNA——Antisense DNA/RNA are oligonucleotides or analogs that are complementary to specific sequences of DNA/RNA. Antisense therapy is an effective treatment, and an antisense drug for viral retinitis has been approved by the FDA [5]. RNAi is a phenomenon of gene silencing that causes the degradation of mRNA. Specific double-stranded RNA (dsRNA) is processed into sRNA, which bind to a kind of proteins to become RNA-induced silencing complex (RISC) that act on mRNA [6-8]. Inserting the siRNA into a cell inhibits targeting mRNA coding, which is an effective strategy for targeting specific diseases. This method is more convenient than knock-out/in, is not affected by the specific structure inside the protein, and costs less.
Pathways——The ways that control and regulate the synthesis of substances in an organism, called Pathways, can help define upstream and downstream targets [9]. Inhibiting or enhancing the synthesis of substances in a specific target pathway increases the efficacy of the drug while reducing side effects, and sometimes even provides a new therapeutic idea. A disease target will have multiple alternative drug targets in the same pathway, resulting in more therapeutic options, for example, based on Jak/Stat Pathway, a drug developed to treat tumors due to chronic inflammation [10].
The number of validated effective drug targets via the methods above remains large. Therefore, further screening of the targets is a very necessary step.
2.2. Screening
High-throughput screening(HTS)——This method generally uses multi-well assay plates (96-, 384-, 1536-well) for parallel measurements [11], one of the smallest volume assays in 1536-well plates is also known as Ultra High-Throughput Screening (UHTS). Today's HTS generally includes automated or semi-automated liquid handling, sample preparation, and data analysis. HTS/UHTS labs often use robots and the latest testing technology to check readouts. Because HTS/UHTS are strongly influenced by solvent stability, environmental factors (temperature, etc.) and statistics (signal-to-noise ratios, Z and Z' quality measures), they will eventually be designed according to their different lower-throughput formats [12].
CADD and SBDD——Development of computer power and X-ray crystallography have also led to the phenomenal growth of computer-aided drug design (CADD) and structure-based drug design (SBDD), both of which are gradually playing a very important role in drug discovery and target screening. Computer screening of compounds is performed by user-defined selection criteria, which can be as simple as the physicochemical properties of the compound or as complex as a three-dimensional construction of the target protein binding site, including conformational analysis and binding energy [13,14]. Computer-aided screening filters out compounds that have no chance of hitting the target. Obviously, this approach allows researchers to reduce the amount of compound trial and error in traditional experiments.
In Vitro/cell-based——Cellular analysis have a wide range of applications, from toxicity analysis, cell growth monitoring, or more complex genetic assays and gene products control. In contrast, in vitro functional analysis facilitates the detection of cellular functions and the monitoring of biological processes, including changes in cell morphology, migration and apoptosis. In vitro cell culture is more ethical and less costly, so in vitro cells are generally used for high-throughput screening, and for the tens of thousands of data points generated during the search for novel drug molecules [15].
In Vivo/animal models——Animal models can mimic specific features of human diseases. Many transgenic animal models have been successfully created, which have been indelibly useful for human prediction of disease mechanisms and pharmacological development [16]. However, for some diseases, such as hepatitis C, there are still no adequate models. The FDA considers the inadequacy of animal models to be a major obstacle to drug development.
2.3. Assessment
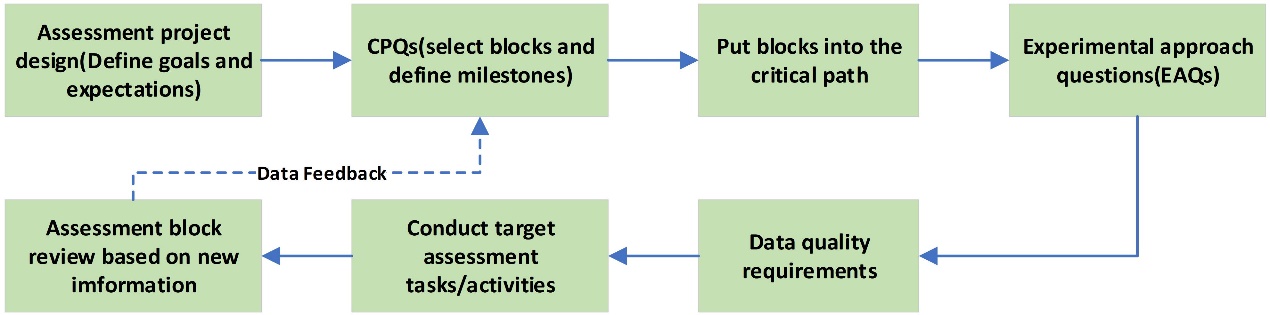
Reliable target evaluation is an important component of drug development, including technical assessment that examine whether the target produce a key effect in the disease process and assessment of whether the target drug is effective in specific diseased populations. As target drugs transition to clinical trials, more issues need to be validated, such as, target availability, potential safety issues, biomarker studies and toxic effects, etc [17]. For target assessment, the GOT-IT (Guidelines On Target Assessment for Innovative Therapeutics) working group has recommendations for target assessment:Drug researchers should raise awareness, prioritize evaluation steps, and use resources effectively in target validation and assessment. Thus, the GOT-IT team recommended grouping target assessment and validation into assessment blocks (AB1-AB5), defining the main framework for mapping the critical path of specific projects. AB1: target-disease linkage; AB2: safety aspects; AB3: microbial targets; AB4: strategic issues (clinical need and commercial potential); AB5: technical feasibility (drug availability, active ingredient determination, etc.), and to develop assessment module tasks, scientists generally use experimental approach questions (EAQs), which can make decisions by using whether the confidence level is met and move to the next step upon success [18]. Figure 1 lists assessment blocks from AB1-AB5.
| |
Figure 1. Assessment blocks from AB1-AB5. | |

At the same time, the selection and ordering of evaluation blocks is determined by critical path questions (CPQs). CPQs help to deepen understanding of the complexity of target assessment and guide scientists in prioritizing assessment blocks in the critical path [19]. While using the CPQs list, scientists can quickly identify obstacles and difficulties in target assessment projects. RNAi drugs are also a type of targeted drug, so they also need to go through the target evaluation mentioned above before they can formally enter the disease treatment research stage. Some CPQs and the steps used by GOT-IT group to define the critical path are listed in Figure 2.
| |
Figure 2. Some CPQs and the steps used by GOT-IT group. | |
2.4. RNAi mechanisms
At the molecular level, mechanisms of RNAi have two common pathways of action, including the miRNA action pathway and the siRNA action pathway.
Pathway of siRNA——In the cytoplasm, exogenous si-RNA and endogenous mi-RNA converge after processing, and the two RNA precursors are sheared by Dicer enzyme into appropriately sized dsRNA that are loaded onto Argonaute proteins. In plant cytoplasm, the Dicer enzyme cleaves first and the 2' hydroxyl group of the 3' nucleotide is then methylated by the methyltransferase HEN1, resulting in a different double-stranded RNA that is subsequently loaded onto the Argonaute protein [20]. The process Dicer enzyme is assisted by dsRNA-binding proteins, such as TARRNA-binding proteins. Three proteins (Dicer, dsRNA binding protein and Argonaute) form a RLC (minimal RISC complex), which is responsible for integrating the cleaved dsRNA and loading it onto Argonaute. dsRNA binds to the PAZ and MID structural domains of the Argonaute protein to generate RISC, one strand of which binds directly to the target RNA to achieve silencing, and the other strand is abandoned. The two chains are called the guide strand and the passenger strand respectively [21]. The transient complex consisting of the Argonaute protein bound to the guide strand and the passenger strand that has not yet been cleaved is called pre-RISC.
Pathway of miRNA——Pri-miRNA (transcripts are at least 1000nt long) in the nucleus are processed by DGCR8 and Dorsha complexes, undergo shearing to form pre-miRNAs, and enter the cytoplasm under the action of transporter enzymes. In the cytoplasm, the processed miRNA is confluent with exogenous siRNA. Dicer enzymes shear miRNA with the help of dsRNA-binding proteins such as TARRNA to form dsRNA of appropriate size and bind to Argonaute proteins to form RISC, the RNA minimal silencing-inducing complex [22]. Under the miRNA pathway, the most common guide strand is called miRNA, while the other strand is miRNA*.
Mechanisms of RNAi drug——Double-stranded RNA (siRNA) causes the termination of translation and gene silencing by breaking down into single-stranded RNA (ssRNA) and integrating into the target mRNA sequence and causing degradation of the target mRNA. Specifically, siRNA drugs cleave double-stranded RNA to produce small molecules of siRNA with the help of RNA endonucleases (helicase) and enter the cytoplasm to start the siRNA action pathway [23]. The siRNA integrates with the protein complex as RISC and forms a coding strand (sense strand) and a template strand (antisense strand). The coding strand is then expelled from the complex and the template strand functions in the complex to process the target mRNA sequence, resulting in gene silencing and inability to synthesize the disease-causing protein [24]. Treatment or prevention of specific diseases through direct inhibition of mRNA expression of disease-causing genes, or inhibition of key receptors (substances) in the pathway of expression of disease-causing genes, making it impossible for viruses to multiply. Numerous protein complexes play an extremely critical role in the functioning of RNAi drugs. Table 1 lists the common protein complexes in the pathways.
Table 1. Common protein complexes in the pathways.
Complex name | Complex function |
DGCR8 Dorsha complex(Microprocessor) | Process pri-miRNA into pre-miRNA |
Dicer family enzymes | Shear miRNAs and siRNAs into the appropriate dsRNA |
Argonaute family proteins | Bind dsRNA to achieve gene silencing of proteins |
RISC | Interference with the RNA silencing induction complex |
RLC | Minimum unit of RISC |
2.5. siRNA-based drug type
By the end of December, 2021, there are four marketed siRNA-based therapeutics: patisiran, givosiran, lumasiran and inclisiran. Besides, six other drugs are in clinical trials, including vutrisiran, nedosiran, fitusiran, teprasiran, cosdosiran and tivanisiran [25,26]. Each siRNA drug treats the specific rare disease, so each has its own specific pathway of action, but all inhibit the production of disease-causing substances through gene silencing. Currently, about 6 indications have been evaluated for the use of siRNA drugs, and many more are still in clinical trials, such as: hemophilia A and hemophilia B (fitusiran), primary angle glaucoma (cosdosiran), ocular pain and dry eye disease (tivanisiran) [27]. Table 2 lists the siRNA drugs that have been put into use and their corresponding disease indications for treatment.
Table 2. siRNA drugs and disease indications.
siRNA-based drug | Indications |
Patisiran | Polyneuropathy due to hereditary transthyretin amyloidosis (hATTR) |
Givosiran | Treatment of acute hepatic porphyria (AHP) in adults |
Lumasiran | Treatment of primary hyperoxaluria type I (PH1), and reduced urinary oxalate levels in pediatric and adult subjects |
Inclisiran | Treatment of adult subjects with heterozygous familial hypercholesterolemia (HeFH), and clinical atherosclerotic cardiovascular disease (ASCVD) |
2.6. Mechanism of siRNA-based drug
Patisiran——It mainly acts against abnormal amyloid TTR formation, so the drug silences the synthetic abnormal amyloid TTR gene [28]. Patisiran is able to bind specifically to genetically conserved sequences of the mRNA of TTR and therefore specifically silences the expression of TTR and inhibits the production of TTR protein, thereby reducing the accumulation of amyloid deposits in peripheral nerves and avoiding organ and tissue damage.
Givosiran——The main target is the defective synthesis of porphyrins, which leads to the accumulation of porphyrin precursors ALA and PGB, causing the patient to develop toxic symptoms. The researchers found that the target ALAS1 plays a key role in the synthesis of ALA and PGB, so they used siRNA drugs to selectively silence mRNA at the ALAS1 locus, thereby reducing ALA and PGB synthesis.
Lumasiran——The reduction of GO enzyme synthesis, and thus oxalate levels, is mainly achieved by silencing the substrate glyoxylate synthase gene and breaking down the hydroxy acid oxidase 1 (HAO1) mRNA. At the same time low oxalate levels lead to less alanine glyoxylate aminotransferase (AGT) and reduce the PH1 pathological mutation process [29].
Inclisiran——This kind of drug couples a double-stranded siRNA targeting PCSK9 to GalNAc targeting hepatocytes, which can be specifically delivered to the target while having high stability. It can degrade the PCSK9 mRNA. The specific mechanism is that PCSK9 disrupts the LDL receptor, but inclisiran inhibits this process, while enhancing the expression of the LDL-C receptor and promoting receptor recycling, making it more susceptible to being hydrolyzed by lysosomes after binding to LDL-C [30]. This leads to an increased uptake of LDL-C by the body, which reduces the level of LDL-C in the blood. Essentially, inclisiran siliences the expression of PCSK9 gene at its source.
2.7. Delivery system
Lipid nanoparticles (LNPs)——Packaging of siRNA into lipid nanoparticles prevents its degradation by in vivo nucleases. Once inside the liver cells, LNP releases its internal siRNA. Because of its lipid composition, it can pass through the cell membrane by endocytosis. It is mainly used for patisiran delivery.
GalNAc conjugates——GalNAc is a ligand that recognizes and binds to a cell surface protein, the desialic acid glycoprotein receptor (ASGPR), which is abundantly expressed on hepatocytes. If several GalNAc units are combined to form a multivalent ligand, the relative binding affinity (RBA) to the receptor increases exponentially. The system is currently available for givosiran, lumasiran and inclisiran administration.
CNS/Ocular-targeted siRNA conjugates——Delivers drugs to the central nervous system and to the eyes.
3. Conclusion
It is an extremely difficult process for targeted drugs to be developed and put into first-line use. The discovery of a disease target may be accidental or based on extensive experiments, and there is no doubt that no matter how the disease target is discovered, the process requires significant human and material resources. Not to mention that the study also has to require the target validation, extensive target screening and target evaluation. Nevertheless, it also demonstrates the tremendous research value of targeted drugs, which offer hope for the treatment of some rare diseases and the possibility of attacking cancers and certain genetic diseases that have plagued humanity for a long time. RNAi drugs have more obvious advantages than other types of drugs, as they can treat diseases at the molecular level, using gene regulation and other means, therefore have fewer side effects on the human body when used correctly. And since the modified dsRNA and protein complexes bind specifically to mRNA, the drugs have high targeting, strong action on specific diseases and better efficacy. However, the drawbacks of RNAi drugs are also a matter of concern for many scientists. RNAi drugs have poor chemical stability. siRNA which is a ribonucleic acid inevitably encounters numerous nucleic acid hydrolases during the process of entering the body to produce medicinal effects [31]. If RNAi drug is not packaged, it may be hydrolyzed and lose its efficacy. Secondly, the negatively charged RNA has difficulty passing through the cell membrane to enter the inner cell for action, which may make the bioavailability of RNAi drugs significantly reduced. The toxic side effects of RNAi drugs cannot be ignored, for example, the sense strand will pair with homologous genes to silence them, thus triggering sense strand-mediated off-target effects, and RNAi drugs may induce immune reactions in vivo. Therefore, improvements to the carriers and delivery system of RNAi drugs are extremely necessary and are key factors in the success of RNAi drugs.
4. Challenges and future perspectives
RNAi-based drug therapy is a creative thought for targeted drug design and has good prospects for development. If scientists can improve the chemical stability of RNAi drugs and the delivery system, it will be a means to treat many diseases. Currently, RNAi drugs are in a booming phase of development and numerous disease indications are being evaluated in clinical trials. The SARS-CoV that is ravaging the world is an RNA virus, and RNAi drug therapy will be an important part of the fight against the virus.
References
[1]. Bolten, B.M. and DeGregorio, T. (2002) Trends in development cycles. Nature Reviews Drug Discovery 1, 335–336.
[2]. Sams-Dodd, Frank (2005) Target-based drug discovery: is something wrong? Drug Discovery Today 10, 139–147.
[3]. FDA (2004) Innovation and Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products. FDA White Paper.
[4]. Dowden, H. & Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov.18, 495–496 (2019).
[5]. Gashaw, I., Ellinghaus, P., Sommer, A. & Asadullah, K. What makes a good drug target? Drug Discov. Today 16, 1037–1043 (2011).
[6]. Everett, J. R. Academic drug discovery: current status and prospects. Expert Opin. Drug Discov. 10, 937–944 (2015).
[7]. Yu, H. W. H. Bridging the translational gap: collaborative drug development and dispelling the stigma of commercialization. Drug Discov. Today 21, 299–305 (2016).
[8]. Moore, J. D. The impact of CRISPR–Cas9 on target identification and validation. Drug Discov. Today 20, 450–457 (2015).
[9]. Erdogan, B. R. & Michel, M. C. Building robustness into translational research. Handb. Exp. Pharmacol. 257, 1–13 (2019).
[10]. Dolgos, H. et al. Translational Medicine Guide transforms drug development processes: the recent Merck experience. Drug Discov. Today 21, 517–526 (2016).
[11]. Andrade, E. L. et al. Non-clinical studies required for new drug development — part I: early in silico and in vitro studies, new target discovery and validation, proof of principles and robustness of animal studies. Braz. J. Med. Biol. Res. 49, e5644 (2016).
[12]. Sweis, R. F. Target (in)validation: a critical, sometimes unheralded, role of modern medicinal chemistry. ACS Med. Chem. Lett. 6, 618–621 (2015).
[13]. Jasny, B. R. et al. Fostering reproducibility in industry–academia research. Science 357, 759–761 (2017).
[14]. Sansone, S.-A. et al. FAIRsharing as a community approach to standards, repositories and policies. Nat. Biotechnol. 37, 358–367 (2019).
[15]. Rattan, A. K. Data integrity: history, issues, and remediation of issues. PDA J. Pharm. Sci. Technol. 72, 105–116 (2018).
[16]. Hartung, T. et al. Toward good in vitro reporting standards. ALTEX 36, 3–17 (2019).
[17]. Berezikov, E. Nat. Rev. Genet. Vol. 12. Nature Publishing Group; 2011. Evolution of microRNA diversity and regulation in animals; p. 846–860
[18]. Emmerich, C. H., Gamboa, L. M., Hofmann, M. C. J., Bonin-Andresen, M., Arbach, O., Schendel, Parnham, M. J. (2020). Improving target assessment in biomedical research: the GOT-IT recommendations. Nature Reviews Drug Discovery.
[19]. Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nature Structural & Molecular Biology. 2012; 19(6):586–593.
[20]. Faller M, Toso D, Matsunaga M, Atanasov I, Senturia R, Chen Y, Zhou ZH, Guo F. DGCR8 recognizes primary transcripts of microRNAs through highly cooperative binding and formation of higher-order structures. RNA. 2010; 16(8):1570–1583.
[21]. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010; 466(7308):835–840.
[22]. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120(1):15–20.
[23]. Zhang MM, Bahal R, Rasmussen TP, Manautou JE, Zhong XB. The growth of siRNA-based therapeutics: Updated clinical studies. Biochem Pharmacol. 2021 Jul;189:114432.
[24]. Senturia R, Faller M, Yin S, Loo JA, Cascio D, Sawaya MR, Hwang D, Clubb RT, Guo F. Structure of the dimerization domain of DiGeorge critical region 8. Protein Sci. 2010; 19(7):1354–1365.
[25]. Tian Y, Simanshu DK, Ascano M, Diaz-Avalos R, Park AY, Juranek SA, Rice WJ, et al. Multimeric assembly and biochemical characterization of the Trax-translin endonuclease complex. Nature Structural & Molecular Biology. 2011; 18(6):658–664.
[26]. Zeng Y, Cullen BR. Efficient processing of primary microRNA hairpins by Drosha requires flanking nonstructured RNA sequences. J. Biol. Chem. 2005; 280(30):27595–27603.
[27]. Zou J, Chang M, Nie P, Secombes CJ. Origin and evolution of the RIG-I like RNA helicase gene family. BMC Evol. Biol. 2009; 9:85.
[28]. Yang J. Patisiran for the treatment of hereditary transthyretin-mediated amyloidosis. Expert Rev Clin Pharmacol. 2019 Feb;12(2):95–99.
[29]. Syed YY. Givosiran: A Review in Acute Hepatic Porphyria. Drugs. 2021 May;81(7):841-848.
[30]. Rizk M, Tüzmen Ş. Update on the clinical utility of an RNA interference-based treatment:focus on Patisiran. Pharmgenomics Pers Med. 2017;10:267–278.
[31]. Xu J, Liu Y, Liu S, Ou W, White A, Stewart S, Tkaczuk KHR, Ellis LM, Wan J, Lu X, HeX. Metformin Bicarbonate-Mediated Efficient RNAi for Precise Targeting of TP53 Deficiency in Colon and Rectal Cancers. Nano Today. 2022 Apr;43
Cite this article
Zhu,W. (2023). RNA interference: from target validation to therapeutics. Theoretical and Natural Science,6,57-64.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the International Conference on Modern Medicine and Global Health (ICMMGH 2023)
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Bolten, B.M. and DeGregorio, T. (2002) Trends in development cycles. Nature Reviews Drug Discovery 1, 335–336.
[2]. Sams-Dodd, Frank (2005) Target-based drug discovery: is something wrong? Drug Discovery Today 10, 139–147.
[3]. FDA (2004) Innovation and Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products. FDA White Paper.
[4]. Dowden, H. & Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov.18, 495–496 (2019).
[5]. Gashaw, I., Ellinghaus, P., Sommer, A. & Asadullah, K. What makes a good drug target? Drug Discov. Today 16, 1037–1043 (2011).
[6]. Everett, J. R. Academic drug discovery: current status and prospects. Expert Opin. Drug Discov. 10, 937–944 (2015).
[7]. Yu, H. W. H. Bridging the translational gap: collaborative drug development and dispelling the stigma of commercialization. Drug Discov. Today 21, 299–305 (2016).
[8]. Moore, J. D. The impact of CRISPR–Cas9 on target identification and validation. Drug Discov. Today 20, 450–457 (2015).
[9]. Erdogan, B. R. & Michel, M. C. Building robustness into translational research. Handb. Exp. Pharmacol. 257, 1–13 (2019).
[10]. Dolgos, H. et al. Translational Medicine Guide transforms drug development processes: the recent Merck experience. Drug Discov. Today 21, 517–526 (2016).
[11]. Andrade, E. L. et al. Non-clinical studies required for new drug development — part I: early in silico and in vitro studies, new target discovery and validation, proof of principles and robustness of animal studies. Braz. J. Med. Biol. Res. 49, e5644 (2016).
[12]. Sweis, R. F. Target (in)validation: a critical, sometimes unheralded, role of modern medicinal chemistry. ACS Med. Chem. Lett. 6, 618–621 (2015).
[13]. Jasny, B. R. et al. Fostering reproducibility in industry–academia research. Science 357, 759–761 (2017).
[14]. Sansone, S.-A. et al. FAIRsharing as a community approach to standards, repositories and policies. Nat. Biotechnol. 37, 358–367 (2019).
[15]. Rattan, A. K. Data integrity: history, issues, and remediation of issues. PDA J. Pharm. Sci. Technol. 72, 105–116 (2018).
[16]. Hartung, T. et al. Toward good in vitro reporting standards. ALTEX 36, 3–17 (2019).
[17]. Berezikov, E. Nat. Rev. Genet. Vol. 12. Nature Publishing Group; 2011. Evolution of microRNA diversity and regulation in animals; p. 846–860
[18]. Emmerich, C. H., Gamboa, L. M., Hofmann, M. C. J., Bonin-Andresen, M., Arbach, O., Schendel, Parnham, M. J. (2020). Improving target assessment in biomedical research: the GOT-IT recommendations. Nature Reviews Drug Discovery.
[19]. Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nature Structural & Molecular Biology. 2012; 19(6):586–593.
[20]. Faller M, Toso D, Matsunaga M, Atanasov I, Senturia R, Chen Y, Zhou ZH, Guo F. DGCR8 recognizes primary transcripts of microRNAs through highly cooperative binding and formation of higher-order structures. RNA. 2010; 16(8):1570–1583.
[21]. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010; 466(7308):835–840.
[22]. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120(1):15–20.
[23]. Zhang MM, Bahal R, Rasmussen TP, Manautou JE, Zhong XB. The growth of siRNA-based therapeutics: Updated clinical studies. Biochem Pharmacol. 2021 Jul;189:114432.
[24]. Senturia R, Faller M, Yin S, Loo JA, Cascio D, Sawaya MR, Hwang D, Clubb RT, Guo F. Structure of the dimerization domain of DiGeorge critical region 8. Protein Sci. 2010; 19(7):1354–1365.
[25]. Tian Y, Simanshu DK, Ascano M, Diaz-Avalos R, Park AY, Juranek SA, Rice WJ, et al. Multimeric assembly and biochemical characterization of the Trax-translin endonuclease complex. Nature Structural & Molecular Biology. 2011; 18(6):658–664.
[26]. Zeng Y, Cullen BR. Efficient processing of primary microRNA hairpins by Drosha requires flanking nonstructured RNA sequences. J. Biol. Chem. 2005; 280(30):27595–27603.
[27]. Zou J, Chang M, Nie P, Secombes CJ. Origin and evolution of the RIG-I like RNA helicase gene family. BMC Evol. Biol. 2009; 9:85.
[28]. Yang J. Patisiran for the treatment of hereditary transthyretin-mediated amyloidosis. Expert Rev Clin Pharmacol. 2019 Feb;12(2):95–99.
[29]. Syed YY. Givosiran: A Review in Acute Hepatic Porphyria. Drugs. 2021 May;81(7):841-848.
[30]. Rizk M, Tüzmen Ş. Update on the clinical utility of an RNA interference-based treatment:focus on Patisiran. Pharmgenomics Pers Med. 2017;10:267–278.
[31]. Xu J, Liu Y, Liu S, Ou W, White A, Stewart S, Tkaczuk KHR, Ellis LM, Wan J, Lu X, HeX. Metformin Bicarbonate-Mediated Efficient RNAi for Precise Targeting of TP53 Deficiency in Colon and Rectal Cancers. Nano Today. 2022 Apr;43