







1. Introduction
1.1. Breast cancer
Breast cancer (BC) is projected to be the most common cancer diagnosed in women (31%) and the second leading cause of cancer-related deaths (15%) in 2023 [1]. BC is further subdivided into four subtypes based on the presence or absence of the following biomarkers: hormone receptors (HR), human epidermal receptor 2 (HER2), and Ki-67.
Patients with BC will be further classified as having luminal A, luminal B, non-luminal HER2+, or triple negative.
Luminal A breast cancer is positive for hormone receptors, negative for HER2+, and has low Ki-67 expression.
(HR+/ HER2-/ low Ki-67). Luminal B breast cancer patients are positive for HR and negative for HER2 but have a high Ki-67 expression (HR+/ HER2-/ high Ki-67). Non-luminal HER2+ test positive for HER2 but negative for HR (HR/HER2+), and triple-negative breast cancer patients do not have any of the three biomarkers [2].
Before BC metastasizes, the cancer is still potentially curable. Nonmetastatic BC [make sure not HER2 stats] have a 90% 5-year survival rate and an 84% 10-year survival rate. However, when BC has metastasized to the lymph nodes, the 5-year survival rate decreases to 86%, and the 10-year survival falls to 29%. Also, metastasized HER2+ breast cancer has a poorer prognosis and higher overall reoccurrence rate [1].
This review summarizes the current knowledge on Lapatinib (Tykerb®), an anti-HER2 drug in the present study, in clinical use along with its mechanism of action and clinical significance.
1.2. HER/EGFR proteins
There are four prominent members of the HER/EGFR family: HER-1 (ErbB1), HER-2 (ErbB2), HER-3 (ErbB3), and HER-4 (ErbB4) [3]. HER receptors consist of five regions: the extracellular ligand-binding region, a transmembrane segment, an intracellular extracellular juxtamembrane region, a cytoplasmic kinase domain, and finally, a C-terminal tail that is later phosphorylated when the protein is activated [4]. The extracellular region is further divided into four parts. The first domain is called L1, and the third (L2) is responsible for ligand binding [5].
When ligands bind to the cysteine-rich extracellular ligand binding site of an HER protein, the inactive, symmetric conformation of the protein dissociates, resulting in an active, asymmetric kinase domain. The activated tyrosine kinase domain facilitates either the homodimerization of one HER receptor or heterodimerization between two HER receptors [5]. The HER-2 protein has no activating ligand, but it is hypothesized that the heterodimerization causes the activated state of HER2 with the other members of the HER/EGFR family, including HER1 and HER3 (Fig 1). The dimerization of the protein results in the phosphorylation of tyrosine residues, ultimately activating a series of signaling pathways, including the MAPK (Ras-Raf-ERK), PI3K/Akt, PKC, and STAT pathways (Fig. 1). These pathways are associated with cell proliferation, invasion, angiogenesis, differentiation, and apoptosis. However, the cellular effects of these signaling pathways vary depending on the characteristics of the ligand binding as well as which HER proteins were involved [6].
The involvement of the HER2 protein promotes more excellent ligand binding and stimulation of signaling pathways, specifically the PI3K/Akt pathway, which regulates cell growth and resistance. Furthermore, the dimerization of HER2 mislocalizes p27Kip1, a cell cycle inhibitor, further promoting cell cycle progression [7].
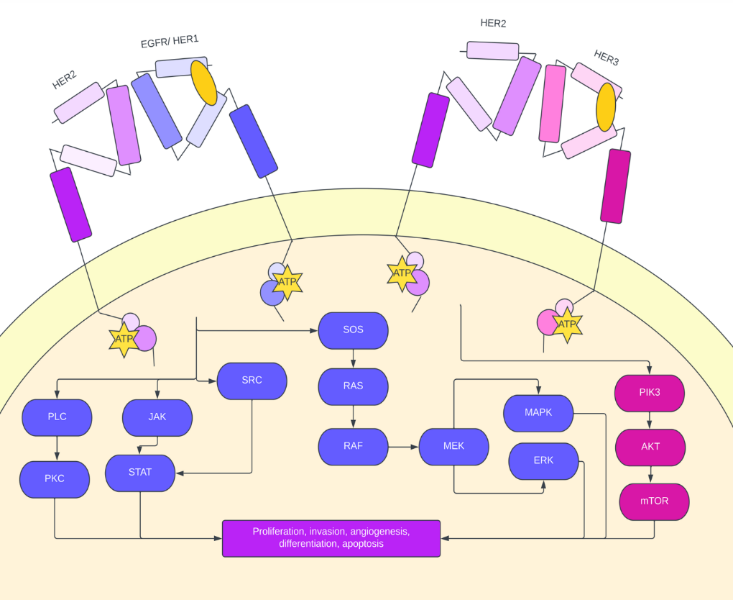
Figure 1. Signaling crosstalk within HER family receptors. Since HER2 has no active ligand binding. site, EGFR (HER1) and HER3 activate the receptor via heterodimerization. HER2 activation through EGFR heterodimerization activates the PLC/PKC, JAK/STAT, and ERK/MAPK pathways, and HER2 activation through HER3 heterodimerization activates the PIK3/AKT pathway. This oncogenic signaling pathway leads to increased proliferation potential, angiogenesis, increased invasion potential, and increased differentiation.
2. HER2 in breast cancer
Approximately 15-20% of breast cancer cases are HER2 positive, as evidenced by HER2 protein overexpression. Patients with breast cancer are tested for HER2 protein positivity by taking a biopsy and conducting either an immunohistochemistry (IHC) test or a fluorescence in situ hybridization (FISH) test [8]. The IHC test measures the percentage of cells out of 100 that stain for hormone receptors and reports a score between 0 and 3. When a patient receives a score of 3 or higher, the patient is HER2+. The IHC test is sometimes followed by the FISH test, which is argued to be more precise but less accessible [9]. The FISH test measures the ratio of HER2 to CEP17. HER2+ is described as a FISH ratio greater than 2.2, equivocal HER2 amplification has a FISH ratio between 1.8 and 2.2, and HER2- is defined as a FISH ratio less than 1.8. Patients with a score of 1 on the IHC test or two on the IHC test with a negative FISH test are considered to have low HER2 breast cancer [10].
Patients with HER2+ breast cancer are more likely to develop brain metastases and have a higher disease recurrence rate. Before BC metastasizes, the tumor is still potentially curable. Nonmetastatic BC has a 90% 5-year survival rate and an 84% 10-year survival rate. However, when BC has metastasized to the lymph nodes, the 5-year survival rate decreases to 86%, and the 10-year survival falls to 29% [1]. HER2+ BC most commonly metastasizes to the bones (65-67% probability), lungs (35-45% probability), and the brain (30-55% probability) [11]. However, the progression-free survival rates (PFS) decreased over the past two decades because of continued biomedical efforts to develop anti-HER2 drug therapies. Trastuzumab is a monoclonal antibody applied as monotherapy or as an adjuvant to chemotherapy. While trastuzumab demonstrates great anticancer and antimetastatic potential, cardiotoxicity is a primary concern, mainly when trastuzumab is used as an adjuvant to anthracycline-based chemotherapy. Patients have also developed a resistance to trastuzumab treatment [12]. Therefore, novel HER2 inhibitors were introduced as alternative forms of treatment.
3. Kinase inhibitors
Tyrosine kinase inhibitors compete with ATP for the binding site of tyrosine kinase to inhibit tyrosine kinase phosphorylation. TKIs are administered orally and generally have low-level adverse effects, including diarrhea. TKIs are highly selective and can either be a mono or dual inhibitor or irreversible or unreversible. To target metastatic HER2+ breast cancer, TKI EGFR inhibitors are administered to prevent or reduce angiogenesis and induce cell-cycle arrest, among other anticancer functions [13]. TKIs are either reversible (Lapatinib, erlotinib, and gefitinib) or irreversible (canertinib, afatinib, and neratinib) [14]. Commonly administered EGFR-TKI inhibitors include Gefitinib and Erlotinib. A brief overview of the mechanism of action, adverse effects, and clinical results are presented below.
Gefitinib, a reversible, antineoplastic EGFR-TKI inhibitor, works to prevent metastasis and angiogenesis and increases the apoptosis of tumor cells. Gefitinib reduces the phosphorylation of basal EGFR, HER2, and HER3. It thus disrupts the formation of the HER3/HER2 heterodimer to activate the HER2 protein [15] (Fig. 2A). Previous studies have also observed that gefitinib increased p27 gene expression and reduced cyclin D and Cdk4 [16] (Fig. 2A). In another study, gefitinib was observed to reduce GSK-3 phosphorylation, which ultimately blocked the AKT signaling pathway. The mTOR signaling pathway is also disrupted by gefitinib [17] (Fig. 2A). Side effects of gefitinib include diarrhea, dry skin, nausea, and vomiting. Results from clinical trials involving gefitinib vary [15]. In one clinical trial that studied HER2+ breast cancer patients (stage IIIb/ IV) with chemotherapy resistance, gefitinib treatment was strongly correlated to reduced EGFR phosphorylation [18]. Polychronis et. al further reports that gefitinib monotherapy reduced tumor size of HER2+ breast cancer patients [19]. Moreover, Kalykaki et. al reported that the number of HER2+ tumor cells was reduced following gefitinib treatment [20]. However, gefitinib demonstrates low anticancer potential in other forms of HER2+ breast cancer. In treating ER+ patients, multiple studies demonstrated the low efficacy of gefitinib in improving clinical benefit [21]. Additionally, for patients previously treated with taxane and anthracycline, gefitinib was shown to be ineffective [15].
Erlotinib, a reversible EGFR-TKI inhibitor, induces cell cycle arrest and apoptosis of cancerous tumor cells. Erlotinib blocks the phosphorylation of the HER2 protein, which ultimately inhibits the MAPK and Akt signaling pathways (Fig 2B). One study reported that erlotinib inhibits HER2 kinase activity by blocking ligand-induced receptor phosphorylation and dimerizing EGFR proteins [22] (Fig 2B). However, erlotinib is not a favored treatment due to its low specificity. In a multicenter phase II trial, erlotinib demonstrated little benefits when given to patients with advanced or metastatic breast cancer [23]. In another clinical trial, Erlotinib effectively treated estrogen receptor-positive breast cancer tumors but not HER2+ breast cancer [24]. Due to erlotinib's lack of specificity, the drug is associated with a higher toxicity level than gefitinib and TKIs with greater specificity. Adverse effects of the drug include grade 3 or 4 nausea (Gefitinib is reported to have a grade level of 1/2 for sickness), diarrhea, vomiting, rash, and acne [22]. Lapatinib, a dual, reversible kinase inhibitor (TKI), competes with ATP for the catalytic kinase domain of HER2/EGFR receptors. By inhibiting the phosphorylation of the receptors, downstream signals are blocked, and cancer behavior-promoting pathways such as the MAPK, AKT, and PI3K pathways are interrupted. Lapatinib demonstrates the most excellent specificity compared to other TKIs, such as afatinib, canertinib, and neratinib [25].
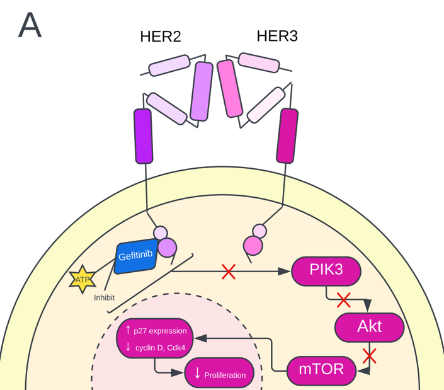
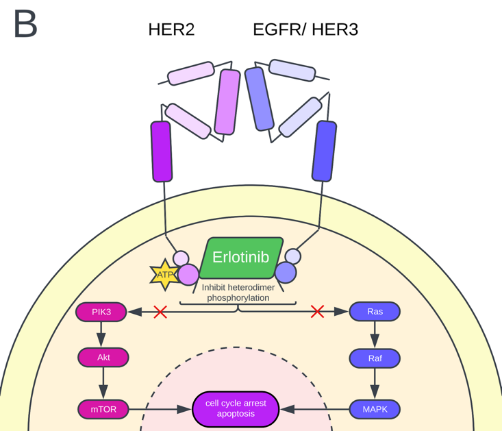
Figure 2. (A) Gefitinib mechanism of action. Gefitinib inhibits the PIK3/Akt pathway, increasing p27 expression and decreasing cyclin D and Cdk4. Ultimately, the proliferation of the cancer cell decreases. (B) Erlotinib, a dual TKI inhibitor, mechanism of action. Erlotinib inhibits the heterodimer phosphorylation of HER2 either by EGFR or HER3. Erlotinib acts on the PIK3/Akt pathway and the MAPK (Ras-Raf-Erk) pathway and ultimately induces cell cycle arrest and apoptosis of the cancer cell.
4. Pharmacology of lapatinib
4.1. Overall structure
Lapatinib is a member of the 4-anilinoquinazoline class of TKIs. The discovery of lapatinib (GW572016, GW2016) resulted from the innovation of bicyclic heteroaromatic compounds as tyrosine kinase inhibitors and knowledge of structure-activity relationships. Lapatinib, developed by Novartis AG, has increased hydrophobic amino groups on the core structure to increase its potency against HER2 and optimize dual ligand binding size to limit pharmacokinetic concerns [26].
4.2. Mechanism of action
Lapatinib competitively inhibits the ATP binding sites found on HER1 and HER2, consequently blocking the phosphorylation of HER1 and HER2. The inability of HER1 and HER2 to phosphorylate then interrupts the MAPK (Raf-Ras-ERK) pathway, AKT, and PI3K pathway, limiting the migration and invasion potential, growth, and proliferation of HER2+ cancer cells [27]. Lapatinib was shown to induce G1 cell cycle arrest in cell lines SNU-216, SNU-484, and NCI-N87. Lapatinib treatment is associated with cMyc reduction and p27kip1 induction, suggesting the ERK pathway is responsible for Lapatinib's inhibitory growth effect on HER2+ cancer cells [29]. The Akt pathway is responsible for Lapatinib's potential to induce apoptosis by inhibiting p-Akt in various cell lines (SNU-216, SNU-484, NCI-N87) and the induction of apoptotic indicators, including caspase three activity (NCI-N87). Lapatinib inhibits nuclear factor-kB activation, consequently blocking the PI3K/ AKT pathway and limiting cancer cells' resistance to radiotherapy [12, 28]. The increase in p38 expression is also observed following Lapatinib treatment. P38, associated with apoptosis and cyclin-dependent kinase inhibitors p21 and p27, will then, therefore, increase the apoptotic potential and the expression of cyclin-dependent kinase inhibitors when the word is increased, resulting in increased apoptosis of cancer cells and limited growth and proliferation of HER2+ cancer cells [30]. Additionally, Lapatinib treatment is associated with increased expression of pro-apoptotic protein BIM and decreased expression of apoptotic inhibitor survivin, thus increasing the apoptotic potential of cancerous HER2+ cells. It is proposed that Lapatinib blocks HER2 and insulin-like growth factor I (IGF-1) cross-talk, resulting in the downregulation of survivin and increased fragmentation of the apoptotic indicator poly-ADP-ribose polymerase (PARP) [32].
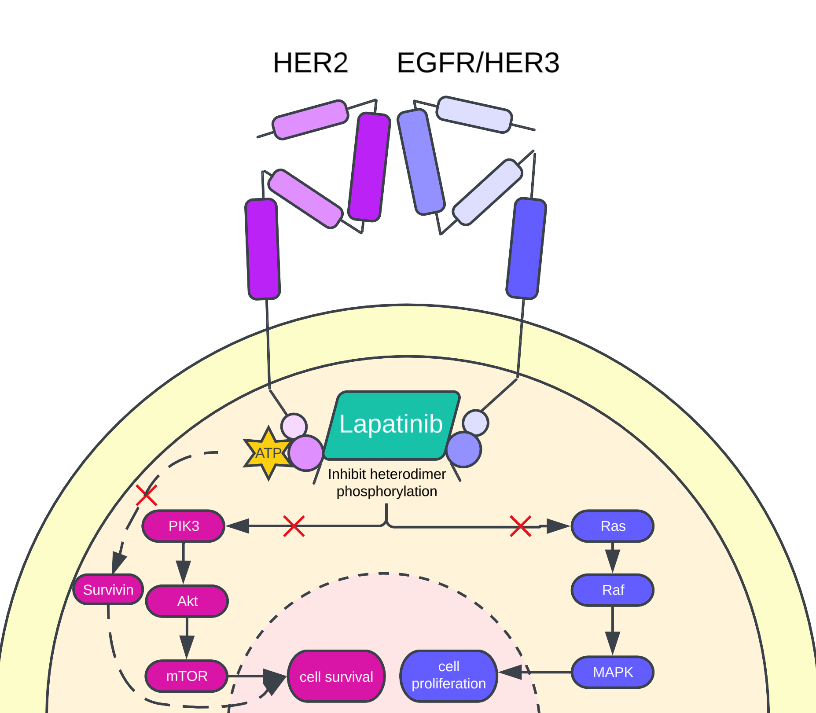
Figure 3. Lapatinib mechanism of action. Lapatinib acts on the PIK3/Akt and the Ras/Raf/MAPK signaling pathway. Additionally, Lapatinib inhibits the expression of apoptotic inhibitor survivin to decrease the cancer cell's survival. Lapatinib reduces cell survival by blocking the PIK3/Akt pathway and cell proliferation by blocking the Ras/Raf/MAPK signaling path.
4.3. Pharmacokinetic properties
The molecular formula of lapatinib is C29H26ClFN4O4S. The approved dose for pharmacokinetic studies ranges from 1,250 mg to 1,500 mg, depending on the combination therapy. Opdam et. al reported a maximum plasma concentration of lapatinib within 3-4 hours following administration and recommended taking lapatinib with a high-fat meal. Joseph et. al corroborates this finding, reporting an AUC increased by 325% following a high-fat meal [32].
Lapatinib has low central nervous system penetration but demonstrates a high affinity to albumin, and α1-acid glycoprotein with more than 99% of lapatinib bounded [36]. Cytochrome CYP3A4 oxidizes lapatinib as shown by an increase in AUC when ketoconazole, a CYP3A4 inhibitor, was administered and a decrease in lapatinib AUC when carbamazepine, a CYP3A4 inducer, was administered [37]. Lapatinib is later eliminated by the liver with a half-life of approximately 14 hours and is more commonly excreted through feces (92%) than urine (<2%) [36].
4.4. Dosage and administration
Lapatinib is commonly administered orally using 250 mg tablets. The recommended daily dose of lapatinib is 1,250 mg for 21 days. However, diarrhea and rash remain the most observed adverse events of lapatinib with amounts up to 1800 mg [35].
It should be noted that for pretreated patients with HER2+ metastatic breast cancer, daily dosage levels from 500 to 1600 were well tolerated. Combined with capecitabine, a lapatinib dose of 1250 mg is recommended for the first 21 days of treatment. An additional 2000 mg of capecitabine is administered daily in twelve-hour intervals for the first 14 days of treatment. Patients should take lapatinib with food that does not include gastric pH-increasing agents and is high in fat or within 30 minutes after food to not intervene with the absorption of the drug [35].
Additionally, given that CYP3A4 metabolizes Lapatinib, it is recommended not to administer lapatinib in addition to CYP3A4 inhibitors or inducers such as Ketoconazole or Carbamazepine, respectively [34].
4.5. Tolerability and toxicity
Diarrhea is the most common form of toxicity observed in HER2+ metastatic breast cancer patients when 500 mg of lapatinib is administered twice daily. Severe diarrhea is observed in approximately 9.7% of patients treated with lapatinib in combination with capecitabine. Other adverse effects of lapatinib and capecitabine combination therapy include hand-foot syndrome, nausea, rash, and fatigue, with most adverse events under the Grade 1 and 2 categories [37]. Cardiotoxicity as an adverse event of lapatinib treatment is not expected as opposed to trastuzumab [38].
4.6. Resistance mechanisms
PI3K/Akt pathway plays a significant role in lapatinib resistance. Expression of FOXO3a, a member of the FOXO subfamily of forehead transcription factors, is regulated by the PI3K/Akt pathway. FoxO proteins are notably involved with apoptosis [40]. Garrett et. al demonstrated that lapatinib-induced HER2 inhibition causes an upregulation of HER3 due to activation of the PI3K/Akt pathway and increasing FOXO3a expression. HER3 upregulation ultimately limits the inhibitory effects of TKIs, and lapatinib resistance is acquired [40].
Additionally, Liu et. al demonstrated that AXL overexpression is associated with acquired resistance to lapatinib, possibly by AXL activating the PI3K pathway [41]. Src tyrosine kinase activity is also related to the PI3K/Akt signaling pathway. Upregulation of Src leads to increased phosphorylation of EGFR, which is suspected of recovering PI3K/Akt signaling inhibited by previous treatment [42]. It is further proposed that mutations in the PI3K/ Akt pathway may lead to lapatinib resistance. Gaining function mutation in PIK3CA genes, notably the upregulation of the P110 protein, triggers lapatinib resistance. However, PIK3CA mutation as a trigger for lapatinib resistance is entirely accepted. In a Japanese clinical study to determine the relationship between PI3K pathway and lapatinib resistance, PIK3CA mutation was only found in 3 of 29 tissue samples [43].
Overexpression of protein tyrosine kinase 6 (PTK6) has been shown to suppress the antigrowth potential of lapatinib. Ito et. al reported the effectiveness of PTK6 inhibitors in overcoming lapatinib resistance in HER2+ breast cancer in vitro [44]. A clinical trial of PTK6 inhibitors in addition to lapatinib treatment was not found. A mutation in the HER2 gene is another proposed mechanism of acquiring lapatinib mutation. Mutants L755S, T7331, T7981, L726F, and T798M induce lapatinib resistance, and a mutation notably at the NH2-terminal kinase lob results in the most excellent lapatinib resistance. EXEL-7647 has been demonstrated to be an effective inhibitor of mutated tumors with lapatinib resistance [46].
4.7. Lapatinib clinical trials
The EMA approved lapatinib use in combination with capecitabine in treating metastatic HER2+ breast cancer due to the results of the EGF100151 clinical trial. The phase III randomized trial assessed the survival, response time, safety, tumor response rate, and clinical benefit rates after the combination therapy. 342 women with HER2+ advanced breast cancer participated in the trial, and each woman was randomly assigned in a 1:1 ratio to receive combination therapy or monotherapy. The most common adverse effect of the combination therapy was diarrhea, with a total of 98 affected of the 342 participants with nausea following the second (72 affected patients). The response rate of the combination therapy group was more significant than the response rate of the monotherapy group (22% vs. 14%, respectively), and the clinical benefit rates were also higher for the combination therapy group (27% vs. 18%). The use of combination therapy reduced the risk of HER2+ breast cancer progression by 51% and further slowed the progression from 4.4 months (monotherapy) to 8.4 months (combination therapy). The trial facilitated further research on lapatinib's use in early HER2+ breast cancer treatment [48]. Gui et. al corroborates these findings through a real-world study by investigating the medical records of 92 patients with HER2+ metastatic breast cancer treated with lapatinib and capecitabine. The study reports an overall survival of 21.5 months, a clinical benefit rate of 47.8%, and an overall progression-free survival of 5.8 months, varying based on the site of metastases. It was further reported that lapatinib with capecitabine treatment performed better than lapatinib chemotherapy treatment in terms of progression-free survival and response rate. Little adverse effects were also observed, making lapatinib capecitabine a promising form of combination therapy [47].
Recently, the use of lapatinib and capecitabine was challenged by the Phase III NALA Trial concerning brain metastases. Neratinib, another TKI, in combination with capecitabine, displayed fever interventions for central ner7ous system diseases, and the duration of response, when treated with neratinib, was longer than lapatinib treatment. However, more significant adverse events were reported when patients were treated with neratinib, and an insignificant change in response rate was reported [48].
4.8. Current Lapatinib Developments
Current research attempts to identify derivatives of Lapatinib with greater affinity for the ATP binding regions of the EGFR and HER2 receptors to promote more excellent inhibitory activity. Synthesized products that contained a quinoline moiety had an inhibitory range of 98.79-99.34% on the EGFR kinase, and N-(3-Chloro-4-(pyridin-2ylmethoxy)phenyl)-7-methoxy-6-(4-(2-nitro-1H-imidazol-1-yl)butoxy) quinazoline-4-amine (LD1) demonstrated the most significant inhibitory effect on both the EGFR and HER2 kinase. LD1 displayed higher potency, improved selectivity, and more incredible binding energy than lapatinib and other synthesized derivatives. LD1 demonstrates excellent potential as a dual EGFR/HER2 inhibitor and requires further research to identify its anticancer potential in vitro and in vivo [49].
The lapatinib derivative identified as 2i has a shorter methylsulfonyl-ethyl-amino-methyl-furyl group compared to Lapatinib, allowing the compound to attach to more external sites of inactive EGFR/HER2. 2i demonstrated a greater affinity and higher binding energy for inactive EGFR and HER2 compared to Lapatinib and performed similarly to Lapatinib regarding selectivity. Lapatinib and its derivatives have more excellent selectivity for passive stages of EGFR and HER2, highlighting that the inactive steps of EGFR/HER2 should be of focus when studying the selectivity of lapatinib derivatives. The methylsulfonyl-ethyl-amino-methyl-furyl group of lapatinib should also be a future study in synthesizing lapatinib derivatives [50].
5. Conclusion
HER2 amplification is associated with more significant metastatic potential, excellent treatment resistance, and poorer prognosis. The continued development of cancer-targeted therapies contributes significantly to treating HER2+ metastatic breast cancer. Lapatinib, approved by the FDA in 2007, continues to be of study due to its low adverse effects and high specificity despite lapatinib being a drug BC patients have acquired resistance to. It is suggested to study the efficacy of lapatinib with resistance inhibitors and lapatinib derivatives in clinical trials in addition to continued efforts to identify lapatinib mechanisms of resistance and lapatinib derivatives with high antimetastatic/anticancer potential.
References
[1]. R. L. Siegel, K. D. Miller, H. E. Fuchs, and A. Jemal, “Cancer statistics, 2022,” CA: A Cancer Journal for Clinicians, vol. 72, no. 1, pp. 7–33, Jan. 2022, doi: https://doi.org/10.3322/caac.21708.
[2]. Patel, N. Unni, and Y. Peng, “The Changing Paradigm for the Treatment of HER2-Positive Breast Cancer,” Cancers, vol. 12, no. 8, p. 2081, Jul. 2020, doi: https://doi.org/10.3390/cancers12082081.
[3]. J. L. Hsu and M.-C. Hung, “The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer,” Cancer and Metastasis Reviews, vol. 35, no. 4, pp. 575–588, Dec. 2016, doi: https://doi.org/10.1007/s10555-016-9649-6.
[4]. P. Wee and Z. Wang, “Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways,” Cancers, vol. 9, no. 12, p. 52, May 2017.
[5]. E. Purba, E. Saita, and I. Maruyama, “Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The ‘Rotation Model,’” Cells, vol. 6, no. 2, p. 13, Jun. 2017, doi: https://doi.org/10.3390/cells6020013.
[6]. Q. Lv et al., “Molecular Mechanisms and Translational Therapies for Human Epidermal Receptor 2 Positive Breast Cancer,” International Journal of Molecular Sciences, vol. 17, no. 12, p. 2095, Dec. 2016, doi: https://doi.org/10.3390/ijms17122095.
[7]. N. Iqbal and N. Iqbal, “Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications,” Molecular Biology International, vol. 2014, no. 1, pp. 1–9, 2014, doi: https://doi.org/10.1155/2014/852748.
[8]. H. Nitta et al., “The assessment of HER2 status in breast cancer: the past, the present, and the future,” Pathology International, vol. 66, no. 6, pp. 313–324, Apr. 2016, doi: https://doi.org/10.1111/pin.12407.
[9]. J. S. Ross, “Point: Fluorescence In Situ Hybridization Is the Preferred Approach over Immunohistochemistry for Determining HER2 Status,” Clinical Chemistry, vol. 57, no. 7, pp. 980–982, Jul. 2011, doi: https://doi.org/10.1373/clinchem.2010.160762.
[10]. U. Krishnamurti and J. F. Silverman, “HER2 in breast cancer: a review and update,” Advances in Anatomic Pathology, vol. 21, no. 2, pp. 100–107, Mar. 2014, doi: https://doi.org/10.1097/PAP.0000000000000015.
[11]. G. Griguolo, T. Pascual, M. V. Dieci, V. Guarneri, and A. Prat, “Interaction of host immunity with HER2-targeted treatment and tumor heterogeneity in HER2-positive breast cancer,” Journal for ImmunoTherapy of Cancer, vol. 7, no. 1, Mar. 2019, doi: https://doi.org/10.1186/s40425-019-0548-6.
[12]. V. Brower, “Cardiotoxicity Debated for Anthracyclines and Trastuzumab in Breast Cancer,” JNCI: Journal of the National Cancer Institute, vol. 105, no. 12, pp. 835–836, Jun. 2013, doi: https://doi.org/10.1093/jnci/djt161.
[13]. N. Spector, W. Xia, I. El-Hariry, Y. Yarden, and S. Bacus, “HER2 therapy. Small molecule HER-2 tyrosine kinase inhibitors,” Breast Cancer Research, vol. 9, no. 2, Mar. 2007, doi: https://doi.org/10.1186/bcr1652.
[14]. G. Iancu et al., “Tyrosine kinase inhibitors in breast cancer (Review),” Experimental and Therapeutic Medicine, vol. 23, no. 2, p. 114, Feb. 2022, doi: https://doi.org/10.3892/etm.2021.11037.
[15]. M. Segovia-Mendoza, M. E. González-González, D. Barrera, L. Díaz, and R. García-Becerra, “Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of HER2-positive breast cancer: preclinical and clinical evidence,” American journal of cancer research, vol. 5, no. 9, pp. 2531–61, 2015, Available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4633889/
[16]. S. H. Ahn, E.-H. Jeong, T.-G. Lee, S. Y. Kim, H.-R. Kim, and C. H. Kim, “Gefitinib induces cytoplasmic translocation of the CDK inhibitor p27 and its binding to a cleaved intermediate of caspase 8 in non-small cell lung cancer cells,” Cellular Oncology (Dordrecht), vol. 37, no. 5, pp. 377–386, Oct. 2014, doi: https://doi.org/10.1007/s13402-014-0198-0.
[17]. P. Duda et al., “Targeting GSK3 and Associated Signaling Pathways Involved in Cancer,” Cells, vol. 9, no. 5, p. 1110, Apr. 2020, doi: https://doi.org/10.3390/cells9051110.
[18]. J. Baselga et al., “Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer,” Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, vol. 23, no. 23, pp. 5323–5333, Aug. 2005, doi: https://doi.org/10.1200/JCO.2005.08.326.
[19]. A. Polychronis et al., “Preoperative gefitinib versus gefitinib and anastrozole in postmenopausal patients with oestrogen-receptor positive and epidermal-growth-factor-receptor-positive primary breast cancer: a double-blind placebo-controlled phase II randomised trial,” The Lancet Oncology, vol. 6, no. 6, pp. 383–391, Jun. 2005, doi: https://doi.org/10.1016/s1470-2045(05)70176-5.
[20]. A. Kalykaki, S. Agelaki, G. Kallergi, A. Xyrafas, D. Mavroudis, and V. Georgoulias, “Elimination of EGFR-expressing circulating tumor cells in patients with metastatic breast cancer treated with gefitinib,” Cancer Chemotherapy and Pharmacology, vol. 73, no. 4, pp. 685–693, Feb. 2014, doi: https://doi.org/10.1007/s00280-014-2387-y.
[21]. C. L. Arteaga et al., “A Phase I-II Study of Combined Blockade of the ErbB Receptor Network with Trastuzumab and Gefitinib in Patients with HER2 (ErbB2)-Overexpressing Metastatic Breast Cancer,” Clinical Cancer Research, vol. 14, no. 19, pp. 6277–6283, Sep. 2008, doi: https://doi.org/10.1158/1078-0432.ccr-08-0482.
[22]. G. Schaefer, L. Shao, K. Totpal, and R. W. Akita, “Erlotinib Directly Inhibits HER2 Kinase Activation and Downstream Signaling Events in Intact Cells Lacking Epidermal Growth Factor Receptor Expression,” Cancer Research, vol. 67, no. 3, pp. 1228–1238, Feb. 2007, doi: https://doi.org/10.1158/0008-5472.can-06-3493.
[23]. M. N. Dickler, M. A. Cobleigh, K. D. Miller, P. M. Klein, and E. P. Winer, “Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer,” Breast Cancer Research and Treatment, vol. 115, no. 1, pp. 115–121, May 2008, doi: https://doi.org/10.1007/s10549-008-0055-9.
[24]. M. Guix et al., “Short preoperative treatment with erlotinib inhibits tumor cell proliferation in hormone receptor-positive breast cancers,” Journal of clinical oncology, vol. 26, no. 6, pp. 897–906, Feb. 2008, doi: https://doi.org/10.1200/jco.2007.13.5939.
[25]. J. E. Frampton, “Lapatinib,” Drugs, vol. 69, no. 15, pp. 2125–2148, Oct. 2009, doi: https://doi.org/10.2165/11203240-000000000-00000.
[26]. D. W. Rusnak et al., “The Effects of the Novel, Reversible Epidermal Growth Factor Receptor/ErbB-2 Tyrosine Kinase Inhibitor, GW2016, on the Growth of Human Normal and Tumor-derived Cell Lines in Vitro and in Vivo,” Molecular Cancer Therapeutics, vol. 1, no. 2, pp. 85–94, Dec. 2001, Accessed: Feb. 04, 2023. [Online]. Available: https://aacrjournals.org/mct/article/1/2/85/233686/The-Effects-of-the-Novel-Reversible-Epidermal
[27]. J.-C. Xuhong, X.-W. Qi, Y. Zhang, and J. Jiang, “Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer,” American Journal of Cancer Research, vol. 9, no. 10, pp. 2103–2119, Oct. 2019, Available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6834479/
[28]. J. W. Kim et al., “The growth inhibitory effect of lapatinib, a dual inhibitor of EGFR and HER2 tyrosine kinase, in gastric cancer cell lines,” Cancer Letters, vol. 272, no. 2, pp. 296–306, Dec. 2008, doi: https://doi.org/10.1016/j.canlet.2008.07.018.
[29]. B. Gril et al., “Effect of Lapatinib on the Outgrowth of Metastatic Breast Cancer Cells to the Brain,” JNCI Journal of the National Cancer Institute, vol. 100, no. 15, pp. 1092–1103, Jul. 2008, doi: https://doi.org/10.1093/jnci/djn216.
[30]. D. Zhang et al., “Activity of lapatinib is independent of EGFR expression level in HER2-overexpressing breast cancer cells,” Molecular Cancer Therapeutics, vol. 7, no. 7, pp. 1846–1850, Jul. 2008, doi: https://doi.org/10.1158/1535-7163.mct-08-0168.
[31]. R. Nahta, L. X. H. Yuan, Y. Du, and F. J. Esteva, “Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling,” Molecular Cancer Therapeutics, vol. 6, no. 2, pp. 667–674, Feb. 2007, doi: https://doi.org/10.1158/1535-7163.MCT-06-0423.
[32]. H. A. Burris et al., “Phase I Safety, Pharmacokinetics, and Clinical Activity Study of Lapatinib (GW572016), a Reversible Dual Inhibitor of Epidermal Growth Factor Receptor Tyrosine Kinases, in Heavily Pretreated Patients With Metastatic Carcinomas,” Journal of Clinical Oncology, vol. 23, no. 23, pp. 5305–5313, Aug. 2005, doi: https://doi.org/10.1200/jco.2005.16.584.
[33]. J. W. Polli et al., “An Unexpected Synergist Role of P-Glycoprotein and Breast Cancer Resistance Protein on the Central Nervous System Penetration of the Tyrosine Kinase Inhibitor Lapatinib (N-{3-Chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016),” Drug Metabolism and Disposition, vol. 37, no. 2, pp. 439–442, Feb. 2009, doi: https://doi.org/10.1124/dmd.108.024646.
[34]. D. A. Smith, K. M. Koch, N. Arya, C. J. Bowen, J. M. Herendeen, and A. Beelen, “Effects of ketoconazole and carbamazepine on lapatinib pharmacokinetics in healthy subjects,” British Journal of Clinical Pharmacology, vol. 67, no. 4, pp. 421–426, Apr. 2009, doi: https://doi.org/10.1111/j.1365-2125.2009.03370.x.
[35]. F. L. Opdam, H. Guchelaar, J. H. Beijnen, and J. H. M. Schellens, “Lapatinib for Advanced or Metastatic Breast Cancer,” The Oncologist, vol. 17, no. 4, pp. 536–542, Apr. 2012, doi: https://doi.org/10.1634/theoncologist.2011-0461.
[36]. G. Capri et al., “An open-label expanded access study of lapatinib and capecitabine in patients with HER2-overexpressing locally advanced or metastatic breast cancer,” Annals of Oncology, vol. 21, no. 3, pp. 474–480, Mar. 2010, doi: https://doi.org/10.1093/annonc/mdp373.
[37]. “European Medicines Agency Tyverb® Summary of Product Characteristics.” Accessed: Feb. 04, 2023. [Online]. Available: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000795/WC500044957.pdf.
[38]. H. D. Choi and M. J. Chang, “Cardiac toxicities of lapatinib in patients with breast cancer and other HER2-positive cancers: a meta-analysis,” Breast Cancer Research and Treatment, vol. 166, no. 3, pp. 927–936, Dec. 2017, doi: https://doi.org/10.1007/s10549-017-4460-9.
[39]. V. D’Amato et al., “Mechanisms of lapatinib resistance in HER2-driven breast cancer,” Cancer Treatment Reviews, vol. 41, no. 10, pp. 877–883, Dec. 2015, doi: https://doi.org/10.1016/j.ctrv.2015.08.001.
[40]. J. T. Garrett et al., “Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase,” Proceedings of the National Academy of Sciences of the United States of America, vol. 108, no. 12, pp. 5021–5026, Mar. 2011, doi: https://doi.org/10.1073/pnas.1016140108.
[41]. L. Liu et al., “Novel Mechanism of Lapatinib Resistance in HER2-Positive Breast Tumor Cells: Activation of AXL,” Cancer Research, vol. 69, no. 17, pp. 6871–6878, Aug. 2009, doi: https://doi.org/10.1158/0008-5472.can-08-4490.
[42]. B. N. Rexer et al., “Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition,” Oncogene, vol. 30, no. 40, pp. 4163–4174, Apr. 2011, doi: https://doi.org/10.1038/onc.2011.130.
[43]. B. Xu et al., “Association of phosphatase and tensin homolog low and phosphatidylinositol 3-kinase catalytic subunit alpha gene mutations on outcome in human epidermal growth factor receptor 2-positive metastatic breast cancer patients treated with first-line lapatinib plus paclitaxel or paclitaxel alone,” Breast Cancer Research, vol. 16, no. 4, Jul. 2014, doi: https://doi.org/10.1186/s13058-014-0405-y.
[44]. D. R. Boulbes et al., “HER family kinase domain mutations promote tumor progression and can predict response to treatment in human breast cancer,” Molecular Oncology, vol. 9, no. 3, pp. 586–600, Nov. 2014, doi: https://doi.org/10.1016/j.molonc.2014.10.011.
[45]. D. Cameron, M. Casey, C. Oliva, B. Newstat, B. Imwalle, and C. E. Geyer, “Lapatinib plus capecitabine in women with HER-2-positive advanced breast cancer: final survival analysis of a phase III randomized trial,” The Oncologist, vol. 15, no. 9, pp. 924–934, 2010, doi: https://doi.org/10.1634/theoncologist.2009-0181.
[46]. X. Gui, H. Li, Y. Yan, and R. Zhang, “Efficacy of lapatinib combined with capecitabine in patients with HER2‑positive metastatic breast cancer in a real‑world study,” Oncology Letters, vol. 20, no. 6, pp. 1–1, Oct. 2020, doi: https://doi.org/10.3892/ol.2020.12241.
[47]. C. Saura et al., “Neratinib Plus Capecitabine Versus Lapatinib Plus Capecitabine in HER2-Positive Metastatic Breast Cancer Previously Treated With ≥ 2 HER2-Directed Regimens: Phase III NALA Trial,” Journal of Clinical Oncology, vol. 38, no. 27, pp. 3138–3149, Sep. 2020, doi: https://doi.org/10.1200/jco.20.00147.
[48]. M. F. Rimawi et al., “Multicenter Phase II Study of Neoadjuvant Lapatinib and Trastuzumab With Hormonal Therapy and Without Chemotherapy in Patients With Human Epidermal Growth Factor Receptor 2–Overexpressing Breast Cancer: TBCRC 006,” Journal of Clinical Oncology, vol. 31, no. 14, pp. 1726–1731, May 2013, doi: https://doi.org/10.1200/jco.2012.44.8027.
[49]. A. Elkamhawy et al., “Design, Synthesis, Biological Evaluation, and Molecular Dynamics Studies of Novel Lapatinib Derivatives,” Pharmaceuticals, vol. 16, no. 1, p. 43, Jan. 2023, doi: https://doi.org/10.3390/
[50]. M. Bello, C. Guadarrama-García, and R. A. Rodriguez-Fonseca, “Dissecting the molecular recognition of dual lapatinib derivatives for EGFR/HER2,” Journal of Computer-Aided Molecular Design, vol. 34, no. 3, pp. 293–303, Dec. 2019, doi: https://doi.org/10.1007/s10822-019-00270-4.
Cite this article
Huang,A. (2023). A review of Lapatinib, a tyrosine kinase inhibitor, in HER2+ metastatic breast cancer treatment development. Theoretical and Natural Science,6,147-157.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the International Conference on Modern Medicine and Global Health (ICMMGH 2023)
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. R. L. Siegel, K. D. Miller, H. E. Fuchs, and A. Jemal, “Cancer statistics, 2022,” CA: A Cancer Journal for Clinicians, vol. 72, no. 1, pp. 7–33, Jan. 2022, doi: https://doi.org/10.3322/caac.21708.
[2]. Patel, N. Unni, and Y. Peng, “The Changing Paradigm for the Treatment of HER2-Positive Breast Cancer,” Cancers, vol. 12, no. 8, p. 2081, Jul. 2020, doi: https://doi.org/10.3390/cancers12082081.
[3]. J. L. Hsu and M.-C. Hung, “The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer,” Cancer and Metastasis Reviews, vol. 35, no. 4, pp. 575–588, Dec. 2016, doi: https://doi.org/10.1007/s10555-016-9649-6.
[4]. P. Wee and Z. Wang, “Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways,” Cancers, vol. 9, no. 12, p. 52, May 2017.
[5]. E. Purba, E. Saita, and I. Maruyama, “Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The ‘Rotation Model,’” Cells, vol. 6, no. 2, p. 13, Jun. 2017, doi: https://doi.org/10.3390/cells6020013.
[6]. Q. Lv et al., “Molecular Mechanisms and Translational Therapies for Human Epidermal Receptor 2 Positive Breast Cancer,” International Journal of Molecular Sciences, vol. 17, no. 12, p. 2095, Dec. 2016, doi: https://doi.org/10.3390/ijms17122095.
[7]. N. Iqbal and N. Iqbal, “Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications,” Molecular Biology International, vol. 2014, no. 1, pp. 1–9, 2014, doi: https://doi.org/10.1155/2014/852748.
[8]. H. Nitta et al., “The assessment of HER2 status in breast cancer: the past, the present, and the future,” Pathology International, vol. 66, no. 6, pp. 313–324, Apr. 2016, doi: https://doi.org/10.1111/pin.12407.
[9]. J. S. Ross, “Point: Fluorescence In Situ Hybridization Is the Preferred Approach over Immunohistochemistry for Determining HER2 Status,” Clinical Chemistry, vol. 57, no. 7, pp. 980–982, Jul. 2011, doi: https://doi.org/10.1373/clinchem.2010.160762.
[10]. U. Krishnamurti and J. F. Silverman, “HER2 in breast cancer: a review and update,” Advances in Anatomic Pathology, vol. 21, no. 2, pp. 100–107, Mar. 2014, doi: https://doi.org/10.1097/PAP.0000000000000015.
[11]. G. Griguolo, T. Pascual, M. V. Dieci, V. Guarneri, and A. Prat, “Interaction of host immunity with HER2-targeted treatment and tumor heterogeneity in HER2-positive breast cancer,” Journal for ImmunoTherapy of Cancer, vol. 7, no. 1, Mar. 2019, doi: https://doi.org/10.1186/s40425-019-0548-6.
[12]. V. Brower, “Cardiotoxicity Debated for Anthracyclines and Trastuzumab in Breast Cancer,” JNCI: Journal of the National Cancer Institute, vol. 105, no. 12, pp. 835–836, Jun. 2013, doi: https://doi.org/10.1093/jnci/djt161.
[13]. N. Spector, W. Xia, I. El-Hariry, Y. Yarden, and S. Bacus, “HER2 therapy. Small molecule HER-2 tyrosine kinase inhibitors,” Breast Cancer Research, vol. 9, no. 2, Mar. 2007, doi: https://doi.org/10.1186/bcr1652.
[14]. G. Iancu et al., “Tyrosine kinase inhibitors in breast cancer (Review),” Experimental and Therapeutic Medicine, vol. 23, no. 2, p. 114, Feb. 2022, doi: https://doi.org/10.3892/etm.2021.11037.
[15]. M. Segovia-Mendoza, M. E. González-González, D. Barrera, L. Díaz, and R. García-Becerra, “Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of HER2-positive breast cancer: preclinical and clinical evidence,” American journal of cancer research, vol. 5, no. 9, pp. 2531–61, 2015, Available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4633889/
[16]. S. H. Ahn, E.-H. Jeong, T.-G. Lee, S. Y. Kim, H.-R. Kim, and C. H. Kim, “Gefitinib induces cytoplasmic translocation of the CDK inhibitor p27 and its binding to a cleaved intermediate of caspase 8 in non-small cell lung cancer cells,” Cellular Oncology (Dordrecht), vol. 37, no. 5, pp. 377–386, Oct. 2014, doi: https://doi.org/10.1007/s13402-014-0198-0.
[17]. P. Duda et al., “Targeting GSK3 and Associated Signaling Pathways Involved in Cancer,” Cells, vol. 9, no. 5, p. 1110, Apr. 2020, doi: https://doi.org/10.3390/cells9051110.
[18]. J. Baselga et al., “Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer,” Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, vol. 23, no. 23, pp. 5323–5333, Aug. 2005, doi: https://doi.org/10.1200/JCO.2005.08.326.
[19]. A. Polychronis et al., “Preoperative gefitinib versus gefitinib and anastrozole in postmenopausal patients with oestrogen-receptor positive and epidermal-growth-factor-receptor-positive primary breast cancer: a double-blind placebo-controlled phase II randomised trial,” The Lancet Oncology, vol. 6, no. 6, pp. 383–391, Jun. 2005, doi: https://doi.org/10.1016/s1470-2045(05)70176-5.
[20]. A. Kalykaki, S. Agelaki, G. Kallergi, A. Xyrafas, D. Mavroudis, and V. Georgoulias, “Elimination of EGFR-expressing circulating tumor cells in patients with metastatic breast cancer treated with gefitinib,” Cancer Chemotherapy and Pharmacology, vol. 73, no. 4, pp. 685–693, Feb. 2014, doi: https://doi.org/10.1007/s00280-014-2387-y.
[21]. C. L. Arteaga et al., “A Phase I-II Study of Combined Blockade of the ErbB Receptor Network with Trastuzumab and Gefitinib in Patients with HER2 (ErbB2)-Overexpressing Metastatic Breast Cancer,” Clinical Cancer Research, vol. 14, no. 19, pp. 6277–6283, Sep. 2008, doi: https://doi.org/10.1158/1078-0432.ccr-08-0482.
[22]. G. Schaefer, L. Shao, K. Totpal, and R. W. Akita, “Erlotinib Directly Inhibits HER2 Kinase Activation and Downstream Signaling Events in Intact Cells Lacking Epidermal Growth Factor Receptor Expression,” Cancer Research, vol. 67, no. 3, pp. 1228–1238, Feb. 2007, doi: https://doi.org/10.1158/0008-5472.can-06-3493.
[23]. M. N. Dickler, M. A. Cobleigh, K. D. Miller, P. M. Klein, and E. P. Winer, “Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer,” Breast Cancer Research and Treatment, vol. 115, no. 1, pp. 115–121, May 2008, doi: https://doi.org/10.1007/s10549-008-0055-9.
[24]. M. Guix et al., “Short preoperative treatment with erlotinib inhibits tumor cell proliferation in hormone receptor-positive breast cancers,” Journal of clinical oncology, vol. 26, no. 6, pp. 897–906, Feb. 2008, doi: https://doi.org/10.1200/jco.2007.13.5939.
[25]. J. E. Frampton, “Lapatinib,” Drugs, vol. 69, no. 15, pp. 2125–2148, Oct. 2009, doi: https://doi.org/10.2165/11203240-000000000-00000.
[26]. D. W. Rusnak et al., “The Effects of the Novel, Reversible Epidermal Growth Factor Receptor/ErbB-2 Tyrosine Kinase Inhibitor, GW2016, on the Growth of Human Normal and Tumor-derived Cell Lines in Vitro and in Vivo,” Molecular Cancer Therapeutics, vol. 1, no. 2, pp. 85–94, Dec. 2001, Accessed: Feb. 04, 2023. [Online]. Available: https://aacrjournals.org/mct/article/1/2/85/233686/The-Effects-of-the-Novel-Reversible-Epidermal
[27]. J.-C. Xuhong, X.-W. Qi, Y. Zhang, and J. Jiang, “Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer,” American Journal of Cancer Research, vol. 9, no. 10, pp. 2103–2119, Oct. 2019, Available: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6834479/
[28]. J. W. Kim et al., “The growth inhibitory effect of lapatinib, a dual inhibitor of EGFR and HER2 tyrosine kinase, in gastric cancer cell lines,” Cancer Letters, vol. 272, no. 2, pp. 296–306, Dec. 2008, doi: https://doi.org/10.1016/j.canlet.2008.07.018.
[29]. B. Gril et al., “Effect of Lapatinib on the Outgrowth of Metastatic Breast Cancer Cells to the Brain,” JNCI Journal of the National Cancer Institute, vol. 100, no. 15, pp. 1092–1103, Jul. 2008, doi: https://doi.org/10.1093/jnci/djn216.
[30]. D. Zhang et al., “Activity of lapatinib is independent of EGFR expression level in HER2-overexpressing breast cancer cells,” Molecular Cancer Therapeutics, vol. 7, no. 7, pp. 1846–1850, Jul. 2008, doi: https://doi.org/10.1158/1535-7163.mct-08-0168.
[31]. R. Nahta, L. X. H. Yuan, Y. Du, and F. J. Esteva, “Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling,” Molecular Cancer Therapeutics, vol. 6, no. 2, pp. 667–674, Feb. 2007, doi: https://doi.org/10.1158/1535-7163.MCT-06-0423.
[32]. H. A. Burris et al., “Phase I Safety, Pharmacokinetics, and Clinical Activity Study of Lapatinib (GW572016), a Reversible Dual Inhibitor of Epidermal Growth Factor Receptor Tyrosine Kinases, in Heavily Pretreated Patients With Metastatic Carcinomas,” Journal of Clinical Oncology, vol. 23, no. 23, pp. 5305–5313, Aug. 2005, doi: https://doi.org/10.1200/jco.2005.16.584.
[33]. J. W. Polli et al., “An Unexpected Synergist Role of P-Glycoprotein and Breast Cancer Resistance Protein on the Central Nervous System Penetration of the Tyrosine Kinase Inhibitor Lapatinib (N-{3-Chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016),” Drug Metabolism and Disposition, vol. 37, no. 2, pp. 439–442, Feb. 2009, doi: https://doi.org/10.1124/dmd.108.024646.
[34]. D. A. Smith, K. M. Koch, N. Arya, C. J. Bowen, J. M. Herendeen, and A. Beelen, “Effects of ketoconazole and carbamazepine on lapatinib pharmacokinetics in healthy subjects,” British Journal of Clinical Pharmacology, vol. 67, no. 4, pp. 421–426, Apr. 2009, doi: https://doi.org/10.1111/j.1365-2125.2009.03370.x.
[35]. F. L. Opdam, H. Guchelaar, J. H. Beijnen, and J. H. M. Schellens, “Lapatinib for Advanced or Metastatic Breast Cancer,” The Oncologist, vol. 17, no. 4, pp. 536–542, Apr. 2012, doi: https://doi.org/10.1634/theoncologist.2011-0461.
[36]. G. Capri et al., “An open-label expanded access study of lapatinib and capecitabine in patients with HER2-overexpressing locally advanced or metastatic breast cancer,” Annals of Oncology, vol. 21, no. 3, pp. 474–480, Mar. 2010, doi: https://doi.org/10.1093/annonc/mdp373.
[37]. “European Medicines Agency Tyverb® Summary of Product Characteristics.” Accessed: Feb. 04, 2023. [Online]. Available: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000795/WC500044957.pdf.
[38]. H. D. Choi and M. J. Chang, “Cardiac toxicities of lapatinib in patients with breast cancer and other HER2-positive cancers: a meta-analysis,” Breast Cancer Research and Treatment, vol. 166, no. 3, pp. 927–936, Dec. 2017, doi: https://doi.org/10.1007/s10549-017-4460-9.
[39]. V. D’Amato et al., “Mechanisms of lapatinib resistance in HER2-driven breast cancer,” Cancer Treatment Reviews, vol. 41, no. 10, pp. 877–883, Dec. 2015, doi: https://doi.org/10.1016/j.ctrv.2015.08.001.
[40]. J. T. Garrett et al., “Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase,” Proceedings of the National Academy of Sciences of the United States of America, vol. 108, no. 12, pp. 5021–5026, Mar. 2011, doi: https://doi.org/10.1073/pnas.1016140108.
[41]. L. Liu et al., “Novel Mechanism of Lapatinib Resistance in HER2-Positive Breast Tumor Cells: Activation of AXL,” Cancer Research, vol. 69, no. 17, pp. 6871–6878, Aug. 2009, doi: https://doi.org/10.1158/0008-5472.can-08-4490.
[42]. B. N. Rexer et al., “Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition,” Oncogene, vol. 30, no. 40, pp. 4163–4174, Apr. 2011, doi: https://doi.org/10.1038/onc.2011.130.
[43]. B. Xu et al., “Association of phosphatase and tensin homolog low and phosphatidylinositol 3-kinase catalytic subunit alpha gene mutations on outcome in human epidermal growth factor receptor 2-positive metastatic breast cancer patients treated with first-line lapatinib plus paclitaxel or paclitaxel alone,” Breast Cancer Research, vol. 16, no. 4, Jul. 2014, doi: https://doi.org/10.1186/s13058-014-0405-y.
[44]. D. R. Boulbes et al., “HER family kinase domain mutations promote tumor progression and can predict response to treatment in human breast cancer,” Molecular Oncology, vol. 9, no. 3, pp. 586–600, Nov. 2014, doi: https://doi.org/10.1016/j.molonc.2014.10.011.
[45]. D. Cameron, M. Casey, C. Oliva, B. Newstat, B. Imwalle, and C. E. Geyer, “Lapatinib plus capecitabine in women with HER-2-positive advanced breast cancer: final survival analysis of a phase III randomized trial,” The Oncologist, vol. 15, no. 9, pp. 924–934, 2010, doi: https://doi.org/10.1634/theoncologist.2009-0181.
[46]. X. Gui, H. Li, Y. Yan, and R. Zhang, “Efficacy of lapatinib combined with capecitabine in patients with HER2‑positive metastatic breast cancer in a real‑world study,” Oncology Letters, vol. 20, no. 6, pp. 1–1, Oct. 2020, doi: https://doi.org/10.3892/ol.2020.12241.
[47]. C. Saura et al., “Neratinib Plus Capecitabine Versus Lapatinib Plus Capecitabine in HER2-Positive Metastatic Breast Cancer Previously Treated With ≥ 2 HER2-Directed Regimens: Phase III NALA Trial,” Journal of Clinical Oncology, vol. 38, no. 27, pp. 3138–3149, Sep. 2020, doi: https://doi.org/10.1200/jco.20.00147.
[48]. M. F. Rimawi et al., “Multicenter Phase II Study of Neoadjuvant Lapatinib and Trastuzumab With Hormonal Therapy and Without Chemotherapy in Patients With Human Epidermal Growth Factor Receptor 2–Overexpressing Breast Cancer: TBCRC 006,” Journal of Clinical Oncology, vol. 31, no. 14, pp. 1726–1731, May 2013, doi: https://doi.org/10.1200/jco.2012.44.8027.
[49]. A. Elkamhawy et al., “Design, Synthesis, Biological Evaluation, and Molecular Dynamics Studies of Novel Lapatinib Derivatives,” Pharmaceuticals, vol. 16, no. 1, p. 43, Jan. 2023, doi: https://doi.org/10.3390/
[50]. M. Bello, C. Guadarrama-García, and R. A. Rodriguez-Fonseca, “Dissecting the molecular recognition of dual lapatinib derivatives for EGFR/HER2,” Journal of Computer-Aided Molecular Design, vol. 34, no. 3, pp. 293–303, Dec. 2019, doi: https://doi.org/10.1007/s10822-019-00270-4.