







1. Introduction
ALS is a motor neuron disease that is very difficult to cure, and it can be easily confused with other diseases in the early stages of its development. ALS is a very scary disease. When it develops, people lose their motor skills gradually and irreversibly. Although people have learned that ALS is caused by damage to motor neurons, current methods can only slow the onset of ALS instead of curing it completely. For example, Riluzole, which is often used in the treatment of ALS, can only prolong the life of ALS patients but does not cure the damage to motor neurons. It is also known through extensive practice that this drug does not work for patients with advanced disease. As a free radical scavenger, Edaravone reduces hydroxyl, hydrogen peroxide, and 3NT levels, but there are strong side effects and it does not work for all kinds of ALS diseases [1]. So, the treatment of ALS can be explored through the study of genes. By exploring how these genes specifically cause the disease in patients, new ways of treating ALS are explored in this paper. Through the investigation of the causative genes, this paper can help readers recognize the danger of ALS and understand its pathogenesis. Moreover, it can also provide some new ideas for ALS treatment.
2. Background information about ALS
The late-onset motor neuron disease ALS is extremely dangerous and deadly. The disease usually occurs in middle age and old age, and the prevalence is higher in men than in women. Lower and upper motor neurons are lost as a result of ALS illness. Upper motor neurons are mainly located in the cortical somatomotor area of the brain, while the cell bodies of lower motor neurons are located in the neuromotor nuclei of the brain and the motor cell sites of the anterior horn of the spinal cord. Due to the damage to upper and lower motor neurons, there is a progressive weakening and atrophy of muscles, including those in the bulb (which are innervated by the medulla oblongata), limbs, trunk, chest, and belly.
3. The effect of the gene TARDBP on ALS
TAR DNA binding protein is the full name of TARDBP. The gene TARDBP is located in 1p36.22. A crucial protein for both sporadic and familial ALS illness is TAR DNA-binding protein 43 (TDP-43), which is encoded by the gene TARDBP. As seen in Figure 1, TDP-43 is mainly found in the nucleus. It is composed of a nuclear export signal, two RNA recognition motifs (RRM1 and RRM2), an N-terminal structural domain, and a C-terminal structural domain made up of TDP-43 protein [2]. However, most of the mutations associated with ALS occur in the C-terminal structural domain. Mutations in the C-terminal gene were found to cause mislocalization of TDP-43 protein from the cytoplasm to the nucleus, resulting in protein truncation and aggregation of the toxic C-terminal TDP-43 fragment and deposition of ubiquitination and hyperphosphorylation forms of TDP-43 [3]. New avenues for the treatment of ALS can be explored through gene therapy for the C-terminus of the mutation and targeted therapy for the mutated gene. A small interfering RNA (siRNA) targeting the TDP-43 mutant allele selectively and preventing the cellular phenotype brought on by the expression of the mutant allele was found to be complementary to the area carrying the point mutation c. 1127G>A [4]. The oxidative stress of the cells was determined by designing comparative experiments to go with fluorescent staining. Using this siRNA to treat the cells that express TDP-43 G376D can greatly reduce the existence of TDP-43 aggregates in the cytoplasm. Although there is no effective way to deliver the siRNA in nerve cells, it can be used as a treatment for neuronal diseases such as ALS. Scientists have discovered a siRNA nano-complex that could be used to treat Alzheimer's disease [5]. Therefore, it is expected that the siRNA can be used to treat ALS disease in a targeted way.
In some TARDBP mutations, it has been found that mutations in genes may affect mitochondrial metabolism and endoplasmic reticulum [6]. To ascertain the impact of the A382T mutation in the TARDBP gene on cell function, researchers analyzed the proteomes of the diseased and wild-typed fibroblasts. Examination of the study's data showed that cytoskeletal dysregulation and impaired mitochondrial activity may be present in ALS fibroblasts. In order to sustain axonal stability, cis- and retrograde transport, and neuronal communication, axonal projections of motor neurons are lengthy and they depend on the health of the neuronal cytoskeleton and mitochondria. Neuronal function is severely impacted by the development of protein aggregates containing components of the cytoskeleton as well as mitochondrial dysfunction [7]. The primary ultrastructural anomalies are found in fibroblasts from ALS patients. The degree of morphological variation in mitochondria is higher; some are noticeably larger with crooked or over-branched cristae, while others have a significant number of decreased cristae. Also, compared to CTRL fibroblasts, endoplasmic reticulum pools in ALS cells are enlarged and irregularly shaped, indicating that the unfolded or misfolded polypeptides are accumulated in the lumen, as seen in Figure 2.
It can be concluded that genetic mutations affect the metabolism and the structure of mitochondria and thus the development of neurons.
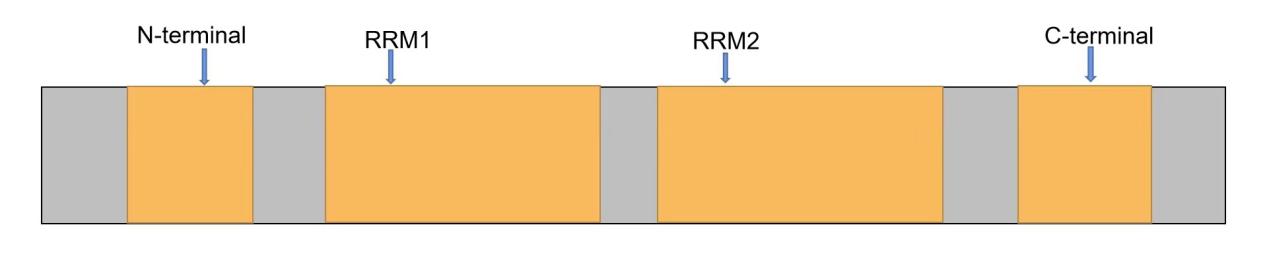
Figure 1. The structure of TAR DNA-binding protein 43 (TDP-43).
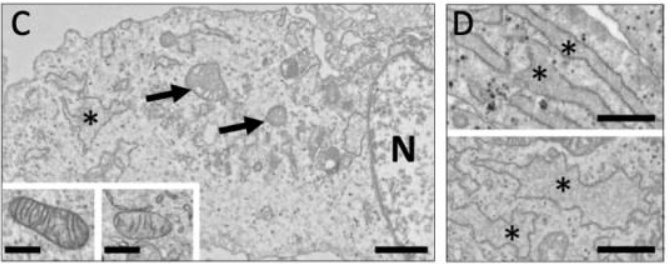
Figure 2. (C) Electron micrographs of fibroblasts from ALS patients.
*Endoplasmic reticulum; N, the nucleus.
A mitochondrion is shown by an arrow. Scale bar: 1 mm. Inserts display a mitochondrion from a control (CTRL) and an ALS fibroblast, respectively (right). 500 nm, insert bar. (D) Endoplasmic reticulum images taken under an electron microscope of CTRL (up) and ALS (down) fibroblasts. Scale bar: 1 mm.
4. The effect of the gene C9ORF72 on ALS
The gene C9ORF72 is also known as the C9orf72-SMCR8 complex subunit. It has been demonstrated that the protein produced by this gene interacts with Rab proteins which are involved in autophagy and endocytic transport, and it plays a significant part in endosomal transport regulation. This gene's transcript has an intron sequence with a GGGGCC repeat that ranges in size from 2-22 to 700-1600 copies and is connected to both 9p-linked ALS and FTD (frontotemporal dementia).
It has been established that dipeptide repeat proteins are produced by the repeat amplification of the C9orf72 gene's ALS-related intron hexanucleotide. Moreover, of dipeptide repeat proteins, polyproline arginine (PR) demonstrates the most potent neurotoxicity both in vitro and in vivo. However, when expressed in neurons and other cells, PR enters the nucleus, and the accumulation of PR in the nucleus interferes with the normal function of the nucleolus. So, reducing the accumulation of PR may reduce the toxicity to neurons [8]. It has been found that the toxic effects on neurons can be reduced by decreasing the level of the transcription factor p53. Nucleoplasmic transport (NCT) deficiency has been suggested as a causative mechanism for repeat amplification toxicity. As a result of the disruption of the Ran-GTPase gradient and NCT due to the deletion of the C9orf72 gene, toxins may accumulate in neurons at a higher rate because of hexanucleotide (GGGGCC) repeat amplification [9]. In the study, it was noted that GGGGCC repeats are able to produce toxic dipeptide repeat proteins (DPRs) independent of the ATG translation promoter, with poly-glycine-alanine repeat polypeptide protein (poly-GA) being the most abundant DPR in patients [10]. However, large amounts of poly-GA may damage the structure of the neuromuscular junction (NMJ) and lead to the development of ALS. By injecting the poly-GA protein into male mice that underwent special treatment, the experimental data can be observed in various ways and poly-GA can affect NMJ to a certain extent. The experimental results show that the poly-GA protein impairs motor function and neurotransmission, diminishes NMJ maintenance, and inhibits aggregation-induced AChR clustering. This study suggests that NMJs can be used as an early therapeutic intervention for ALS and other neuromuscular diseases. Although it remains to be discussed whether in vitro poly-GA affects neurosynaptic transmission in the nervous system, this approach may also be a therapeutic idea for ALS disease.
5. Some methods of gene therapy
In the previous sections, the author described ALS disease caused by the gene TARDBP and C9ORF72. Gene therapy is a very effective treatment for neurodegenerative diseases like ALS. From Figure 3, several gene therapies that are used to treat neurodegenerative diseases can be seen. Antisense oligonucleotides (ASOs) and siRNAs, which are single-stranded deoxyribonucleotide analogs, are the foundation of RNA modification therapies. By controlling protein translation, interacting with RNA, or interfering with the splicing machinery, they can change how genes are expressed. siRNAs are RNA double-stranded bodies that cause gene silencing by containing strands that lead to RNA-induced silencing complexes, called RNA interference. Mutations occurring at the C-terminus of the gene TARDBP can be targeted by siRNA. ASO and siRNA-based RNA modification therapies and viral vectors are two examples of the novel techniques applied in the therapeutic strategies discussed. As a single-stranded DNA virus, AAV is a type of microvirus. After systemic delivery of AAV9, high expression is seen in the cervical spinal cord, hippocampus, motor cortex, and cerebellum. This strongly suggests that this method shows promise in the treatment of neurological diseases [11].
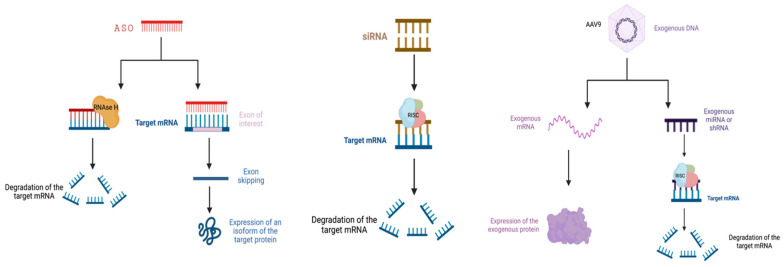
Figure 3. Various methods of gene therapy.
6. Conclusion
This paper discusses the causes of the disease ALS and the influence of genes on ALS. Of the genes that can cause ALS, TARDBP and C9ORF72 are two typical ones. For the gene TARDBP, siRNA targeting can be used to affect the mutation in the C-terminus of the gene. In the case of C9ORF72, the in vitro synthesis of the poly-GA protein can be used to affect NMJ to alleviate and prevent neuromuscular diseases. The author believes that the approach of the gene therapy is necessary for a disease like ALS, although it seems that the gene therapy for ALS has some limitations and uncertainties at present, and it is not mature enough. ALS is a neurological disease that is extremely difficult to cure. Current treatments can only slow down its onset and add survival time for ALS patients even after a long period of exploration. Therefore, for future studies, scientists can increase their research efforts and explore the treatment for ALS in depth from the genetic level.
References
[1]. Neupane, P., Thada, P. K., Singh, P., Faisal, A. R., Rai, N., Poudel, P., Waleed, M. S., Quinonez, J., Ruxmohan, S., Jain, E. Investigating Edaravone Use for Management of Amyotrophic Lateral Sclerosis (ALS): A Narrative Review. Cureus 15(1), e33746 (2023). doi: 10.7759/cureus.33746.
[2]. Pinarbasi, E. S., Cağatay, T., Fung, H. Y. J., Li, Y. C., Chook, Y. M., Thomas, P. J. Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci Rep. 8(1), 7083 (2018). doi: 10.1038/s41598-018-25008-4.
[3]. Suk, T. R., Rousseaux, M. W. C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener 15(1), 45 (2020). doi: 10.1186/s13024-020-00397-1.
[4]. Romano, R., De Luca, M., Del Fiore, V. S., Pecoraro, M., Lattante, S., Sabatelli, M., La Bella, V., Bucci, C. Allele-specific silencing as therapy for familial amyotrophic lateral sclerosis caused by the p.G376D TARDBP mutation. Brain Commun 4(6), fcac315 (2022). doi: 10.1093/braincomms/fcac315.
[5]. Guo, Q., Zheng, X., Yang, P., Pang, X., Qian, K., Wang, P., Xu, S., Sheng, D., Wang, L., Cao, J., Lu, W., Zhang, Q., Jiang, X. Small interfering RNA delivery to the neurons near the amyloid plaques for improved treatment of Alzheimer׳s disease. Acta Pharm Sin B 9(3), 590-603 (2019). doi: 10.1016/j.apsb.2018.12.010.
[6]. Zanini, G., Selleri, V., Nasi, M., De Gaetano, A., Martinelli, I., Gianferrari, G., Lofaro, F. D., Boraldi, F., Mandrioli, J., Pinti, M. Mitochondrial and Endoplasmic Reticulum Alterations in a Case of Amyotrophic Lateral Sclerosis Caused by TDP-43 A382T Mutation. Int J Mol Sci. 23(19), 11881 (2022). doi: 10.3390/ijms231911881.
[7]. Theunissen, F., West, P. K., Brennan, S., Petrović, B., Hooshmand, K., Akkari, P. A., Keon, M., Guennewig, B. New perspectives on cytoskeletal dysregulation and mitochondrial mislocalization in amyotrophic lateral sclerosis. Transl Neurodegener. 10(1), 46 (2021). doi: 10.1186/s40035-021-00272-z.
[8]. Cicardi, M. E., Hallgren, J. H., Mawrie, D., Krishnamurthy, K., Markandaiah, S. S., Nelson, A. T., Kankate, V., Anderson, E. N., Pasinelli, P., Pandey, U. B., Eischen, C. M., Trotti, D. C9orf72 poly (PR) mediated neurodegeneration is associated with nucleolar stress. bioRxiv [Preprint] (2023). doi: 10.1101/2023.02.16.528809.
[9]. McGoldrick, P., Lau, A., You, Z., Durcan, T. M., Robertson, J. Loss of C9orf72 perturbs the Ran-GTPase gradient and nucleocytoplasmic transport, generating compositionally diverse Importin β-1 granules. Cell Rep. 42(3), 112134 (2023). doi: 10.1016/j.celrep.2023.112134.
[10]. Tu, W. Y., Xu, W., Zhang, J., Qi, S., Bai, L., Shen, C., Zhang, K. C9orf72 poly-GA proteins impair neuromuscular transmission. Zool Res. 44(2), 331-340 (2023). doi: 10.24272/j.issn.2095-8137.2022.356.
[11]. Lejman, J., Panuciak, K., Nowicka, E., Mastalerczyk, A., Wojciechowska, K., Lejman, M. Gene Therapy in ALS and SMA: Advances, Challenges and Perspectives. Int J Mol Sci. 24(2), 1130 (2023). doi: 10.3390/ijms24021130.
Cite this article
Zang,J. (2023). Analysis on the effect of genes on amyotrophic lateral sclerosis. Theoretical and Natural Science,6,229-233.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the International Conference on Modern Medicine and Global Health (ICMMGH 2023)
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Neupane, P., Thada, P. K., Singh, P., Faisal, A. R., Rai, N., Poudel, P., Waleed, M. S., Quinonez, J., Ruxmohan, S., Jain, E. Investigating Edaravone Use for Management of Amyotrophic Lateral Sclerosis (ALS): A Narrative Review. Cureus 15(1), e33746 (2023). doi: 10.7759/cureus.33746.
[2]. Pinarbasi, E. S., Cağatay, T., Fung, H. Y. J., Li, Y. C., Chook, Y. M., Thomas, P. J. Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci Rep. 8(1), 7083 (2018). doi: 10.1038/s41598-018-25008-4.
[3]. Suk, T. R., Rousseaux, M. W. C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener 15(1), 45 (2020). doi: 10.1186/s13024-020-00397-1.
[4]. Romano, R., De Luca, M., Del Fiore, V. S., Pecoraro, M., Lattante, S., Sabatelli, M., La Bella, V., Bucci, C. Allele-specific silencing as therapy for familial amyotrophic lateral sclerosis caused by the p.G376D TARDBP mutation. Brain Commun 4(6), fcac315 (2022). doi: 10.1093/braincomms/fcac315.
[5]. Guo, Q., Zheng, X., Yang, P., Pang, X., Qian, K., Wang, P., Xu, S., Sheng, D., Wang, L., Cao, J., Lu, W., Zhang, Q., Jiang, X. Small interfering RNA delivery to the neurons near the amyloid plaques for improved treatment of Alzheimer׳s disease. Acta Pharm Sin B 9(3), 590-603 (2019). doi: 10.1016/j.apsb.2018.12.010.
[6]. Zanini, G., Selleri, V., Nasi, M., De Gaetano, A., Martinelli, I., Gianferrari, G., Lofaro, F. D., Boraldi, F., Mandrioli, J., Pinti, M. Mitochondrial and Endoplasmic Reticulum Alterations in a Case of Amyotrophic Lateral Sclerosis Caused by TDP-43 A382T Mutation. Int J Mol Sci. 23(19), 11881 (2022). doi: 10.3390/ijms231911881.
[7]. Theunissen, F., West, P. K., Brennan, S., Petrović, B., Hooshmand, K., Akkari, P. A., Keon, M., Guennewig, B. New perspectives on cytoskeletal dysregulation and mitochondrial mislocalization in amyotrophic lateral sclerosis. Transl Neurodegener. 10(1), 46 (2021). doi: 10.1186/s40035-021-00272-z.
[8]. Cicardi, M. E., Hallgren, J. H., Mawrie, D., Krishnamurthy, K., Markandaiah, S. S., Nelson, A. T., Kankate, V., Anderson, E. N., Pasinelli, P., Pandey, U. B., Eischen, C. M., Trotti, D. C9orf72 poly (PR) mediated neurodegeneration is associated with nucleolar stress. bioRxiv [Preprint] (2023). doi: 10.1101/2023.02.16.528809.
[9]. McGoldrick, P., Lau, A., You, Z., Durcan, T. M., Robertson, J. Loss of C9orf72 perturbs the Ran-GTPase gradient and nucleocytoplasmic transport, generating compositionally diverse Importin β-1 granules. Cell Rep. 42(3), 112134 (2023). doi: 10.1016/j.celrep.2023.112134.
[10]. Tu, W. Y., Xu, W., Zhang, J., Qi, S., Bai, L., Shen, C., Zhang, K. C9orf72 poly-GA proteins impair neuromuscular transmission. Zool Res. 44(2), 331-340 (2023). doi: 10.24272/j.issn.2095-8137.2022.356.
[11]. Lejman, J., Panuciak, K., Nowicka, E., Mastalerczyk, A., Wojciechowska, K., Lejman, M. Gene Therapy in ALS and SMA: Advances, Challenges and Perspectives. Int J Mol Sci. 24(2), 1130 (2023). doi: 10.3390/ijms24021130.