







1. Introduction
Protein aggregation diseases are a category of disorders characterized by the misfolding of specific proteins and the formation of abnormal aggregates that subsequently trigger cytotoxic responses [1]. Protein aggregation serves as a critical pathological hallmark in a group of neurodegenerative diseases (NDs), including the formation of amyloid-β (Aβ) plaques and Tau neurofibrillary tangles (NFTs) in Alzheimer’s disease (AD), as well as the aggregation of α-synuclein (α-Syn) in Parkinson’s disease (PD), Parkinson’s disease-related dementia (PDD), and multiple system atrophy (MSA). A common pathological feature among these diseases is the disruption of proteostasis, leading to the self-assembly of soluble protein monomers into oligomers, protofibrils, and ultimately insoluble amyloid fibrils. Based on the deposition sites of pathological proteins, protein aggregation diseases can be classified into two major categories: systemic amyloidoses (such as light-chain amyloidosis and transthyretin amyloidosis) and neurodegenerative diseases (including AD, PD, and Huntington’s disease) [1]. Although the clinical manifestations of these diseases vary, they share several features: the conformational transition and self-aggregation of specific proteins, the cytotoxicity of pathological aggregates, and the presence of a “prion-like” cell-to-cell transmission mechanism.
Selecting AD as a paradigm for studying protein aggregation diseases is supported by both scientific rationale and practical necessity. Its significance lies in its dual value as both a representative example and an entry point for understanding similar disorders. AD exemplifies the core features and complexity of protein aggregation diseases. Its key pathological proteins, Aβ and Tau, represent two classical modes of protein aggregation: Aβ aggregation occurs primarily extracellularly, forming characteristic amyloid plaques—analogous to the pathological process of many systemic amyloidoses—whereas Tau aggregation occurs intracellularly within neurons, forming NFTs. This makes AD an excellent model for studying the mechanisms and consequences of intracellular protein aggregation [2]. Thus, AD constitutes a comprehensive research system encompassing both extracellular and intracellular protein aggregation, and its study provides valuable insights for understanding the broader class of these diseases. Moreover, the high global prevalence of AD underscores the urgency of research and its immense translational medical value. According to epidemiological data, AD affects tens of millions of people worldwide, and there are currently no curative therapies capable of slowing or halting disease progression. This enormous unmet medical need strongly motivates the translation of basic research discoveries into clinical applications. Any breakthrough in AD therapeutic strategies would not only bring hope to countless patients and families but could also offer valuable guidance and technical references for treating other protein aggregation diseases.
In summary, due to AD’s unique pathological complexity, extensive research foundation, and intense research focus, it serves as an ideal model for deepening our understanding of protein aggregation diseases and for developing broadly applicable therapeutic strategies. The innovation of this review lies in its approach of exploring the shared molecular mechanisms underlying protein aggregation diseases, with a particular emphasis on AD as a paradigm. This work examines the intricate pathological network of AD and its exemplary role in the study of this disease category. Such an analysis not only facilitates a comprehensive understanding of the pathological mechanisms of protein aggregation diseases but also provides theoretical support for developing both universal and targeted therapeutic strategies. By integrating current research progress and multidisciplinary perspectives, this review aims to offer new insights for future research and provide a scientific foundation for the development of more effective clinical interventions.
2. The common molecular mechanisms of protein aggregation diseases
Although protein aggregation diseases exhibit diverse clinical manifestations, their pathogenesis shares a set of highly conserved molecular pathways, which includes three common molecular mechanisms: the loss of proteostasis, kinetic of aggregation and toxic species, as well as “prion-like” propagation of pathological proteins.
2.1. Loss of proteostasis
The quality control of intracellular proteins relies on a sophisticated surveillance network known as proteostasis, the failure of which initiates protein aggregation to a certain extent. Cellular machineries for proteostasis include the chaperone system, the ubiquitin–proteasome system (UPS), and the autophagy–lysosome pathway [3].
Molecular chaperones (e.g., heat shock protein 70 and heat shock protein 90) play crucial roles in nascent protein synthesis, folding, and the stress response by recognizing and assisting misfolded proteins in refolding. Under conditions of aging or cellular stress, chaperone function becomes insufficient or overloaded, impairing the effective repair of misfolded proteins and creating favorable conditions for aggregation.
The UPS serves as the primary mechanism for degrading short-lived and misfolded proteins within cells. Misfolded proteins are tagged with ubiquitin and subsequently recognized and degraded by the proteasome. However, abnormally aggregated protein oligomers or fibrils often inhibit proteasomal activity, forming a vicious cycle that further exacerbates the collapse of proteostasis.
The autophagy–lysosome pathway functions as a clearance system for long-lived proteins, protein aggregates, and damaged organelles [3]. When UPS function is impaired or aggregates become too large to be degraded by the proteasome, cells activate autophagy to facilitate clearance. In protein aggregation diseases, autophagic flux is frequently blocked by pathological proteins (e.g., mutant huntingtin or α-synuclein), preventing the effective removal of toxic aggregates and leading to their massive accumulation. The functional decline or overload of these systems collectively contributes to the collapse of the proteostasis network, establishing a common molecular foundation for the onset of various protein aggregation diseases.
2.2. Kinetic of aggregation and toxic species
Rather than a simple precipitation process, protein aggregation is a multistep reaction that follows distinct kinetic phases, progressing from nucleation to elongation [4]. Specifically, nucleation is an energetically unfavorable and slow process in which a few monomers randomly collide to form an unstable oligomeric core. This stage serves as the rate-limiting step of aggregation. Once a stable nucleus is formed, additional monomers rapidly attach to it, leading to fiber elongation, an autocatalytic process. Mature fibrils can then fragment under mechanical stress or enzymatic activity, generating numerous new ends that serve as sites for secondary nucleation, thereby accelerating the aggregation process exponentially. Throughout this process, various aggregate species are formed. Recent studies consistently indicate that soluble oligomers are the most toxic species, as they induce membrane damage and trigger multiple universal mechanisms of cellular toxicity [5].
On one hand, oligomers interact with cell membranes—particularly with lipid rafts—forming nonspecific pores or disrupting membrane integrity, which leads to ion imbalance and cellular leakage. On the other hand, oligomers can target mitochondria, disrupting membrane potential, impairing ATP production, and promoting the excessive generation of reactive oxygen species (ROS). Furthermore, mitochondrial dysfunction and the activation of inflammatory responses collectively trigger a burst of ROS, exacerbating oxidative damage to proteins and further compromising proteostasis. This establishes a positive feedback loop that amplifies cellular damage and accelerates disease progression.
2.3. “Prion-like” propagation of pathological proteins
Pathological protein aggregates are now known to possess the ability to spread between cells through a process termed “prion-like” propagation, which represents a common mechanism underlying the progressive spread of disease within the brain [6].
This process begins with the release of pathological protein aggregates into the extracellular space, which can occur through cell death, exosome secretion, or non-classical secretory pathways. These extracellular aggregates are then taken up by neighboring neurons via endocytosis, receptor-mediated endocytosis, or direct membrane penetration. Once internalized, the exogenous pathological aggregates act as seeds within the recipient cells, recruiting and converting normal proteins into misfolded forms, thereby achieving self-replication.
This mechanism explains why the spatiotemporal progression of many protein aggregation diseases is highly predictable and closely correlates with the worsening of clinical symptoms. Consequently, blocking the cell-to-cell propagation of pathological proteins has emerged as a highly promising therapeutic strategy for halting or slowing disease progression.
3. Specific aggregation mechanisms in AD
AD, as a paradigmatic example of protein aggregation diseases, exhibits both the common molecular mechanisms discussed in the previous section and unique pathological complexities. To comprehensively understand AD, it is essential to explore how these shared molecular features manifest specifically in its pathology, as well as to examine its distinctive disease system, with a particular focus on the pathogenic Aβ and Tau.
3.1. Mechanisms of AD
3.1.1. Production, aggregation and toxicity of Aβ
Aβ peptide is generated through the sequential cleavage of amyloid precursor protein (APP) by β-secretase and γ-secretase [7]. Under physiological conditions, Aβ may have certain normal functions, and its production and clearance remain in dynamic balance. In Alzheimer’s disease (AD), however, this balance becomes disrupted. Genetic studies have shown that mutations in the APP gene or in presenilin (a core component of the γ-secretase complex), which cause familial AD, directly lead to the overproduction of Aβ₄₂, the 42–amino acid isoform of Aβ. Aβ₄₂ has a higher propensity to aggregate, initially forming soluble oligomers. These Aβ oligomers have been demonstrated in numerous studies to be highly synaptotoxic, capable of impairing long-term potentiation, disrupting synaptic plasticity, and activating microglia to trigger inflammatory responses [8]. Ultimately, the oligomers further aggregate into insoluble amyloid plaques that are deposited extracellularly. Although these plaques are less acutely toxic than soluble oligomers, they act as a persistent “reservoir of toxicity”, contributing to chronic neuroinflammation and disrupting brain tissue architecture.
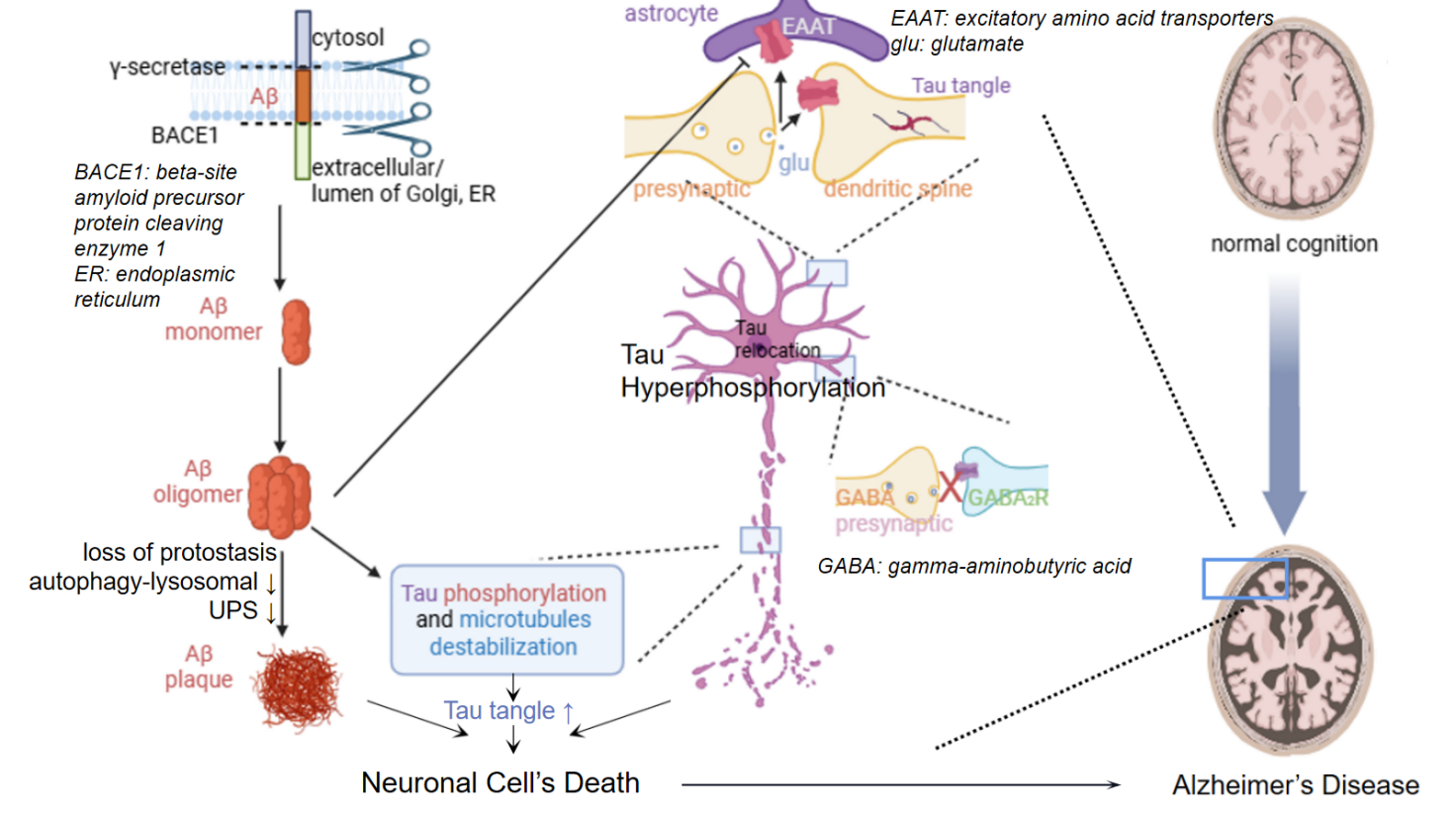
Figure 1 shows the molecular mechanisms of AD. In the amyloid pathway, sequential cleavage by β-amyloid precursor protein cleavage enzyme 1 (BACE1) and γ-secretase generates Aβ monomer, which aggregates into toxic Aβ oligomers and ultimately Aβ plaques. This triggers Tau hyperphosphorylation, leading to microtubule destabilization and Tau tangle formation. These pathologies collectively cause synaptic dysfunction, including disruptions in glutamatergic and GABAergic signaling, and dendritic spine loss. This eventually results in neuronal death and cognitive decline, progressing from normal cognition to AD.
3.1.2. Pathological transformation of Tau
In healthy neurons, Tau protein primarily binds to microtubules, helping stabilize their structure. However, in AD pathology, Tau undergoes abnormal hyperphosphorylation, causing it to detach from microtubules. This detachment leads to a loss of normal function, resulting in decreased microtubule stability and impaired intracellular transport. The detached and misfolded Tau proteins subsequently aggregate into pathological oligomers and eventually form neurofibrillary tangles (NFTs). Through this process, Tau acquires toxic properties. Pathological Tau oligomers can disrupt organelle functions—such as those of mitochondria—and impair synaptic activity. Importantly, the accumulation of NFTs shows a much stronger correlation with neuronal death and cognitive decline than the presence of amyloid plaques [9].
3.1.3. Manifestation of common mechanisms in AD
As an ideal model of protein aggregation diseases, AD serves as a textbook example illustrating the common pathological mechanisms underlying this class of disorders [10]. On one hand, aging and genetic factors contribute to a decline in the neuronal proteostasis network. For instance, reduced UPS function decreases the clearance efficiency of misfolded Aβ and Tau, while impaired autophagy-lysosomal pathway hinders the effective degradation of aggregates, thereby creating conditions conducive to the development of Aβ and Tau pathology (Figure 1).
On the other hand, Aβ and Tau oligomers exhibit distinct but complementary neurotoxic effects. Aβ oligomers primarily target the postsynaptic membrane, disrupting glutamatergic neurotransmission [11], whereas Tau oligomers mainly interfere with axonal transport and neuronal energy metabolism. Acting synergistically, these two toxic species assault neurons from both extracellular and intracellular fronts [12]. Moreover, AD exemplifies the “prion-like” propagation of Tau pathology. Pathological Tau proteins spread through the brain along a highly predictable anatomical pathway—from the entorhinal cortex to the hippocampus, and subsequently to the neocortex [13]. This pattern of dissemination closely mirrors the progression of clinical disease stages, directly explaining the gradual worsening of symptoms observed in AD.
3.2. Complexity and uniqueness of AD
Although the main pathological progression of AD serves as a representative example of common protein aggregation mechanisms, AD is far from being a simple, single-pathway disorder driven solely by Aβ or Tau. Its uniqueness lies in the complex pathological network formed by the interplay of multiple molecular and cellular elements.
The interaction between Aβ and Tau follows a hierarchical pattern in which Aβ acts upstream while Tau functions as a downstream executor. Substantial evidence supports the “Aβ upstream, Tau execution” hypothesis: pathological aggregation of Aβ—particularly in its oligomeric form—serves as a key trigger that accelerates and exacerbates Tau hyperphosphorylation and aggregation. The subsequent spread of Tau pathology, in turn, directly contributes to neuronal death and cognitive decline [12].
Furthermore, the central and chronic role of neuroinflammation in AD pathogenesis is increasingly recognized. Aβ plaques and neuronal damage chronically activate the brain’s innate immune cells, including microglia and astrocytes [14]. Initially, this inflammatory response serves a protective role, aiming to clear Aβ deposits and cellular debris. However, as the disease progresses, the inflammation becomes chronic and dysregulated, leading to the excessive release of pro-inflammatory cytokines that exacerbate neuronal injury and impair repair mechanisms. This creates a vicious cycle, in which inflammation becomes an independent driver of disease progression [15].
In addition, genetic risk factors play a crucial role in modulating AD pathogenesis. For instance, the ε4 allele of the apolipoprotein E (ApoE) gene is recognized as the strongest genetic risk factor for AD. Dysfunction of the ApoE4 isoform impairs Aβ clearance, promotes its aggregation, disrupts synaptic plasticity, and enhances neuroinflammation [16]. These findings underscore the profound interconnections between lipid metabolism, innate immunity, and protein aggregation in AD, highlighting the intricate and multifactorial nature of its pathological mechanisms.
4. Universal strategies and the AD example
Building upon a deep understanding of the molecular mechanisms underlying protein aggregation diseases, the development of therapeutic strategies is advancing at an unprecedented pace. In this chapter, various mechanism-based therapeutic strategies are systematically elaborated, with a particular focus on AD, to explore their applications, challenges, and prospects in a specific disease context. By targeting the core steps of protein aggregation, these strategies aim to reduce the production of toxic species at their source.
4.1. Universal strategies targeting aggregation process and their application in AD
4.1.1. Inhibit production of Aβ
This strategy aims to reduce the source production of the pathogenic protein. In AD, BACE-1 inhibitors were once considered highly promising, as they are designed to block Aβ generation. BACE-1 performs a crucial, rate-limiting step by cleaving APP, producing soluble APPβ and a membrane-anchored C-terminal fragment called C99. C99 is then processed by γ-secretase, resulting in the formation of neurotoxic Aβ. Consequently, the development of selective and potent BACE-1 inhibitors represents a highly promising therapeutic approach for AD [17]. Despite their significant potential, the clinical progression of many early BACE-1 inhibitors was hindered by challenges such as poor brain penetration, insufficient selectivity, and undesirable side effects [18]. Nevertheless, these efforts validated the rationale of targeting protein production as a universal therapeutic strategy.
4.1.2. Inhibit aggregation of misfolded proteins
This approach, also known as “Promote Disassembly,” aims to prevent misfolded proteins from assembling into toxic forms or even to disassemble pre-formed aggregates. In AD, small-molecule Tau aggregation inhibitors—such as methylthioninium chloride (MTC) and its derivatives, including leuco-methylthioninium bis (LMTM)—are under clinical investigation [19]. Tau aggregation is a self-propagating process that begins when Tau binds to a non-specific substrate, exposing a domain that enables Tau proteins to bind each other with high affinity. The initial formation of this substrate complex is likely triggered by an age-related decline in endosomal-lysosomal system function, which is responsible for processing cellular proteins, particularly mitochondrial membrane proteins. In individuals with mutations in APP or the presenilin complex, this age-related failure of protein processing is exacerbated, accelerating the onset of the Tau aggregation cascade rather than directly causing it [20]. These compounds bind to Tau, preventing the formation of β-sheet structures and thereby inhibiting oligomer and fibril formation [21]. This strategy holds broad-spectrum potential, applicable to various protein aggregation diseases.
4.1.3. Enhance clearance of protein aggregates
This method leverages endogenous or exogenous mechanisms to clear existing protein aggregates. Immunotherapy has emerged as a leading strategy in this area. In Alzheimer’s disease, monoclonal antibodies against Aβ, such as Aducanumab and Lecanemab, have been approved for early-stage patients, promoting microglia-mediated clearance of Aβ [22]. Similarly, antibodies targeting pathological Tau are under clinical investigation, aiming to block cell-to-cell transmission and facilitate clearance [23]. These successes highlight the immense potential of immunotherapy as a cross-disease, universal platform. However, challenges remain in optimizing efficacy and managing side effects: randomized clinical trials have identified a potential adverse event known as amyloid-related imaging abnormalities, detectable via magnetic resonance imaging and occurring spontaneously or as a treatment-related effect [24].
4.1.4. Boost autophagy cellular clearance of protein aggregates
This strategy enhances the cell’s intrinsic “waste disposal” systems to remove toxic aggregates. Autophagy inducers, such as Rapamycin and its analogs, have shown promise in preclinical models by clearing both Aβ and Tau aggregates, representing an attractive universal approach [25]. However, safely and effectively modulating autophagy in humans remains a significant challenge.
4.2. Strategies targeting downstream pathological events
When directly targeting aggregated proteins proves challenging, intervening in the downstream pathological events they trigger becomes an important complementary approach. Chronic neuroinflammation is a common driver in protein aggregation diseases. In AD, anti-neuroinflammatory strategies—such as targeting the triggering receptor expressed on myeloid cells 2 (TREM2) on microglia—have gained significant attention. TREM2 agonists aim to shift harmful inflammatory states toward protective ones, enhancing Aβ clearance and supporting neuronal health [26]. This strategy is broadly applicable to neurological diseases where protein aggregation induces substantial inflammation.
In addition, neuroprotection and metabolic modulation aim to enhance neuronal resilience to aggregate-induced toxicity or improve brain energy metabolism. Approaches include mitochondrial protectors, antioxidants, and existing symptomatic drugs such as acetylcholinesterase inhibitors, Memantine, and multi-target-directed ligands. These compounds offer distinct advantages by not only enhancing the therapeutic efficacy of single-target agents through synergistic effects but also mitigating potential side effects [27]. While they do not alter disease progression, they exemplify the universal value of symptomatic and supportive treatments by providing relief and supporting neuronal function, making them indispensable in the management of protein aggregation diseases.
4.3. Emerging platform technologies
With the rapid advancement of scientific research, cutting-edge biotechnologies are providing revolutionary tools for treating Alzheimer’s disease and other protein aggregation disorders. Gene therapy is one such approach. These methods employ technologies such as antisense oligonucleotides (ASOs) or small interfering RNA to reduce the expression of pathogenic proteins at the mRNA level. In AD, Tau-targeting ASOs are being investigated to decrease the production of all Tau isoforms at the source, thereby preventing the formation and spread of pathological Tau [28].
Stem cell therapy represents another promising strategy. It aims either to replace lost cells by transplanting stem cells or their differentiated neurons, or to provide neurotrophic support by transplanting cells such as neural progenitor cells to create a protective environment [29]. Although technical challenges remain, the combined potential for cell replacement and support offers hope for patients in advanced disease stages.
Finally, precision medicine and early intervention have become cornerstones for the success of all these strategies. Using blood and cerebrospinal fluid biomarkers, such as p-tau181 and p-tau217, along with positron emission tomography (PET) imaging, clinicians can identify at-risk individuals years or even decades before clinical symptoms emerge [30]. This represents a fundamental shift from reactive disease treatment to biomarker-driven early intervention, providing the opportunity to apply disease-modifying therapies before irreversible damage occurs.
5. Conclusion
Protein aggregation diseases share a core set of molecular pathogenic mechanisms, centered on the loss of proteostasis, the formation of toxic oligomers, and their cell-to-cell propagation. AD, a representative example, serves as an ideal paradigm for elucidating these mechanisms. It not only exemplifies the common processes of Aβ and Tau protein aggregation but also reveals their complex interactions, along with the unique pathological networks formed by neuroinflammation and genetic factors. Consequently, effective therapeutic strategies for protein aggregation diseases require a dual approach: developing broad-spectrum therapies targeting universal steps in protein aggregation and designing precise interventions aimed at AD-specific targets such as Aβ and Tau.
Despite significant progress in understanding these diseases, challenges remain, including identifying the precise molecular triggers of aggregation and addressing the inherent complexity of these pathological networks. Future research should focus on improving our understanding of early disease markers and developing targeted therapies capable of intervening before irreversible damage occurs. Additionally, leveraging emerging technologies—such as gene editing, precision medicine, and advanced imaging—could open new avenues for both diagnosis and treatment. Translating preclinical findings into effective clinical therapies remains a crucial yet challenging step.
Ultimately, in-depth research on AD and other protein aggregation diseases holds significance beyond any single disorder. Insights gained from AD research will provide critical clues for the prevention and treatment of the broader spectrum of protein aggregation diseases and drive overall progress in neurodegenerative disease research.
References
[1]. Wu, J., Wu, J., Chen, T., Cai, J., and Ren, R. (2024) Protein aggregation and its affecting mechanisms in neurodegenerative diseases. Neurochemistry International, 180, 105880.
[2]. Zheng, Q., and Wang, X. (2025) Alzheimer’s disease: insights into pathology, molecular mechanisms, and therapy. Protein & Cell, 16(2), 83-120.
[3]. Fan, T., Peng, J., Liang, H., Chen, W., Wang, J., and Xu, R. (2025) Potential common pathogenesis of several neurodegenerative diseases. Neural Regeneration Research, 21(3), 972-988.
[4]. Sun, K. T., and Mok, S. -A. (2025) Inducers and modulators of protein aggregation in Alzheimer’s disease - Critical tools for understanding the foundations of aggregate structures. Neurotherapeutics, 22(3), e00512.
[5]. Simon, P. Y. R., and David, R. (2025) Alzheimer’s Disease, β-Amyloid Peptides, and Ubiquitin-Proteasome System: Nutritherapeutic Insights. Neurodegenerative Disease, 25(2), 76-87.
[6]. Eid, S., Lee, S., Verkuyl, C. E., Almanza, D., Hanna, J., Shenouda, S., Belotserkovsky, A., Zhao, W., and Watts, J. C. (2024) The importance of prion research. Biochemistry and Cell Biology, 102(6), 448-471.
[7]. Aivalioti, E., Georgiopoulos, G., Tual-Chalot, S., Bampatsias, D., Delialis, D., Sopova, K., Drakos, S. G., Stellos, K., and Stamatelopoulos, K. (2025) Amyloid-beta metabolism in age-related neurocardiovascular diseases. European Heart Journal, 46(3), 250-272.
[8]. Ding, S., Choi, S. H., and Miller, Y. I. (2025) Amyloid β-Induced Inflammarafts in Alzheimer’s Disease. International Journal of Molecular Sciences, 26(10), 4592.
[9]. Zhang, X., Liu, Y., Rekowski, M. J., and Wang, N. (2025) Lactylation of tau in human Alzheimer’s disease brains. Alzheimer’s & Dementia, 21(2), e14481.
[10]. Kamatham, P. T., Shukla, R., Khatri, D. K., and Vora, L. K. (2024) Pathogenesis, diagnostics, and therapeutics for Alzheimer’s disease: Breaking the memory barrier. Ageing Research Reviews, 101, 102481.
[11]. Mishra, P., Esfahani, E. K., Fernyhough, P., and Albensi, B. C. (2025) Estradiol Prevents Amyloid Beta-Induced Mitochondrial Dysfunction and Neurotoxicity in Alzheimer’s Disease via AMPK-Dependent Suppression of NF-κB Signaling. International Journal of Molecular Sciences, 26(13), 6203.
[12]. Kadamangudi, S., Marcatti, M., Zhang, W. -R., Fracassi, A., Kayed, R., Limon, A., and Taglialatela, G. (2024) Amyloid-β oligomers increase the binding and internalization of tau oligomers in human synapses. Acta Neuropathologica, 149(1), 2.
[13]. Balducci, C., Orsini, F., Cerovic, M. et al. (2025) Tau oligomers impair memory and synaptic plasticity through the cellular prion protein. Acta Neuropathologica Communications, 13(1), 17.
[14]. Tiong, S. Q., Mohgan, R. N., Quek, J. Y., Liew, J. Y. S., Wong, G. Y. S., Thang, Z. Q., Chan, Z. L., Gan, S. Y., and Chan, E. W. L. (2025) Inhibition of the Transforming Growth Factor-β Signaling Pathway Confers Neuroprotective Effects on Beta-Amyloid-Induced Direct Neurotoxicity and Microglia-Mediated Neuroinflammation. Neurology Research International, 8948290.
[15]. Zhang, G., Peng, Q., Guo, X. et al. (2025) Microglia-derived Galectin-9 drives amyloid-β pathology in Alzheimer’s disease. Aging Cell, 24(2), e14396.
[16]. Wang, X., Wang, J., He, Y., Li, H., Yuan, H., Evans, A., Yu, X., He, Y., and Wang, H. (2015) Apolipoprotein E ε4 Modulates Cognitive Profiles, Hippocampal Volume, and Resting-State Functional Connectivity in Alzheimer’s Disease. Journal of Alzheimer’s Disease, 45(3), 781-95.
[17]. M, Y., and Shetty, N. S. (2025) Advances in the Synthetic Approaches to β‑Secretase (BACE-1) Inhibitors in Countering Alzheimer’s: A Comprehensive Review. ACS Omega, 10(32), 35367-35433.
[18]. Monteiro, K. L. C., Dos Santos Alcântara, M. G., Freire, N. M. L., Brandão, E. M., do Nascimento, V. L., Dos Santos Viana, L. M., de Aquino, T. M., and da Silva-Júnior, E. F. (2023) BACE-1 Inhibitors Targeting Alzheimer’s Disease. Current Alzheimer Research, 20(3), 131-148.
[19]. Melis, V., Magbagbeolu, M., Rickard, J. E. et al. (2015) Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav Pharmacol. 2015 Jun; 26(4): 353-68.
[20]. Wischik CM, Harrington CR, Storey JM. Tau-aggregation inhibitor therapy for Alzheimer's disease. Behavioural Pharmacology, 88(4), 529-39.
[21]. Duggal, A., Mahindru, D., Baghel, K., Mehrotra, S., and Prajapati, V. K. (2025) Tau protein aggregation: A therapeutic target for neurodegenerative diseases. Advances in Protein Chemistry and Structural Biology, 146, 77-136.
[22]. Jucker, M., and Walker, L. C. (2023) Alzheimer’s disease: From immunotherapy to immunoprevention. Cell, 186(20), 4260-4270.
[23]. Guo, X., Yan, L., Zhang, D., and Zhao, Y. (2024) Passive immunotherapy for Alzheimer’s disease. Ageing Research Reviews, 94, 102192.
[24]. Hampel, H., Elhage, A., Cho, M., Apostolova, L. G., Nicoll, J. A. R., and Atri, A. (2023) Amyloid-related imaging abnormalities (ARIA): radiological, biological and clinical characteristics. Brain, 146(11), 4414-4424.
[25]. Zhang, Z., Yang, X., Song, Y. Q., and Tu, J. (2021) Autophagy in Alzheimer's disease pathogenesis: Therapeutic potential and future perspectives. Ageing Research Reviews, 72, 101464.
[26]. Qin, Q., Teng, Z., Liu, C., Li, Q., Yin, Y., and Tang, Y. (2021) TREM2, microglia, and Alzheimer’s disease. Mechanisms of Ageing and Development, 195, 111438.
[27]. Zhao, X., Hu, Q., Wang, X. et al. (2024) Dual-target inhibitors based on acetylcholinesterase: Novel agents for Alzheimer's disease. European Journal of Medicinal Chemistry, 279, 116810.
[28]. Vemula, P., Schoch, K. M., and Miller, T. M. (2023) Evaluating the efficacy of purchased antisense oligonucleotides to reduce mouse and human tau in vivo. Frontiers in Molecular Neuroscience, 16, 1320182.
[29]. Cao, Z., Kong, F., Ding, J., Chen, C., He, F., and Deng, W. (2024) Promoting Alzheimer’s disease research and therapy with stem cell technology. Stem Cell Research & Therapy, 15(1), 136.
[30]. Wang, J., Jin, C., Zhou, J., Zhou, R., Tian, M., Lee, H. J., and Zhang, H. (2022) PET molecular imaging for pathophysiological visualization in Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging, 50(3), 765-783.
Cite this article
Yang,X. (2025). Protein Aggregation Diseases: Mechanistic Insights and Therapeutic Strategies with a Focus on Alzheimer’s Disease. Theoretical and Natural Science,147,45-54.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of ICBioMed 2025 Symposium: AI for Healthcare: Advanced Medical Data Analytics and Smart Rehabilitation
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Wu, J., Wu, J., Chen, T., Cai, J., and Ren, R. (2024) Protein aggregation and its affecting mechanisms in neurodegenerative diseases. Neurochemistry International, 180, 105880.
[2]. Zheng, Q., and Wang, X. (2025) Alzheimer’s disease: insights into pathology, molecular mechanisms, and therapy. Protein & Cell, 16(2), 83-120.
[3]. Fan, T., Peng, J., Liang, H., Chen, W., Wang, J., and Xu, R. (2025) Potential common pathogenesis of several neurodegenerative diseases. Neural Regeneration Research, 21(3), 972-988.
[4]. Sun, K. T., and Mok, S. -A. (2025) Inducers and modulators of protein aggregation in Alzheimer’s disease - Critical tools for understanding the foundations of aggregate structures. Neurotherapeutics, 22(3), e00512.
[5]. Simon, P. Y. R., and David, R. (2025) Alzheimer’s Disease, β-Amyloid Peptides, and Ubiquitin-Proteasome System: Nutritherapeutic Insights. Neurodegenerative Disease, 25(2), 76-87.
[6]. Eid, S., Lee, S., Verkuyl, C. E., Almanza, D., Hanna, J., Shenouda, S., Belotserkovsky, A., Zhao, W., and Watts, J. C. (2024) The importance of prion research. Biochemistry and Cell Biology, 102(6), 448-471.
[7]. Aivalioti, E., Georgiopoulos, G., Tual-Chalot, S., Bampatsias, D., Delialis, D., Sopova, K., Drakos, S. G., Stellos, K., and Stamatelopoulos, K. (2025) Amyloid-beta metabolism in age-related neurocardiovascular diseases. European Heart Journal, 46(3), 250-272.
[8]. Ding, S., Choi, S. H., and Miller, Y. I. (2025) Amyloid β-Induced Inflammarafts in Alzheimer’s Disease. International Journal of Molecular Sciences, 26(10), 4592.
[9]. Zhang, X., Liu, Y., Rekowski, M. J., and Wang, N. (2025) Lactylation of tau in human Alzheimer’s disease brains. Alzheimer’s & Dementia, 21(2), e14481.
[10]. Kamatham, P. T., Shukla, R., Khatri, D. K., and Vora, L. K. (2024) Pathogenesis, diagnostics, and therapeutics for Alzheimer’s disease: Breaking the memory barrier. Ageing Research Reviews, 101, 102481.
[11]. Mishra, P., Esfahani, E. K., Fernyhough, P., and Albensi, B. C. (2025) Estradiol Prevents Amyloid Beta-Induced Mitochondrial Dysfunction and Neurotoxicity in Alzheimer’s Disease via AMPK-Dependent Suppression of NF-κB Signaling. International Journal of Molecular Sciences, 26(13), 6203.
[12]. Kadamangudi, S., Marcatti, M., Zhang, W. -R., Fracassi, A., Kayed, R., Limon, A., and Taglialatela, G. (2024) Amyloid-β oligomers increase the binding and internalization of tau oligomers in human synapses. Acta Neuropathologica, 149(1), 2.
[13]. Balducci, C., Orsini, F., Cerovic, M. et al. (2025) Tau oligomers impair memory and synaptic plasticity through the cellular prion protein. Acta Neuropathologica Communications, 13(1), 17.
[14]. Tiong, S. Q., Mohgan, R. N., Quek, J. Y., Liew, J. Y. S., Wong, G. Y. S., Thang, Z. Q., Chan, Z. L., Gan, S. Y., and Chan, E. W. L. (2025) Inhibition of the Transforming Growth Factor-β Signaling Pathway Confers Neuroprotective Effects on Beta-Amyloid-Induced Direct Neurotoxicity and Microglia-Mediated Neuroinflammation. Neurology Research International, 8948290.
[15]. Zhang, G., Peng, Q., Guo, X. et al. (2025) Microglia-derived Galectin-9 drives amyloid-β pathology in Alzheimer’s disease. Aging Cell, 24(2), e14396.
[16]. Wang, X., Wang, J., He, Y., Li, H., Yuan, H., Evans, A., Yu, X., He, Y., and Wang, H. (2015) Apolipoprotein E ε4 Modulates Cognitive Profiles, Hippocampal Volume, and Resting-State Functional Connectivity in Alzheimer’s Disease. Journal of Alzheimer’s Disease, 45(3), 781-95.
[17]. M, Y., and Shetty, N. S. (2025) Advances in the Synthetic Approaches to β‑Secretase (BACE-1) Inhibitors in Countering Alzheimer’s: A Comprehensive Review. ACS Omega, 10(32), 35367-35433.
[18]. Monteiro, K. L. C., Dos Santos Alcântara, M. G., Freire, N. M. L., Brandão, E. M., do Nascimento, V. L., Dos Santos Viana, L. M., de Aquino, T. M., and da Silva-Júnior, E. F. (2023) BACE-1 Inhibitors Targeting Alzheimer’s Disease. Current Alzheimer Research, 20(3), 131-148.
[19]. Melis, V., Magbagbeolu, M., Rickard, J. E. et al. (2015) Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav Pharmacol. 2015 Jun; 26(4): 353-68.
[20]. Wischik CM, Harrington CR, Storey JM. Tau-aggregation inhibitor therapy for Alzheimer's disease. Behavioural Pharmacology, 88(4), 529-39.
[21]. Duggal, A., Mahindru, D., Baghel, K., Mehrotra, S., and Prajapati, V. K. (2025) Tau protein aggregation: A therapeutic target for neurodegenerative diseases. Advances in Protein Chemistry and Structural Biology, 146, 77-136.
[22]. Jucker, M., and Walker, L. C. (2023) Alzheimer’s disease: From immunotherapy to immunoprevention. Cell, 186(20), 4260-4270.
[23]. Guo, X., Yan, L., Zhang, D., and Zhao, Y. (2024) Passive immunotherapy for Alzheimer’s disease. Ageing Research Reviews, 94, 102192.
[24]. Hampel, H., Elhage, A., Cho, M., Apostolova, L. G., Nicoll, J. A. R., and Atri, A. (2023) Amyloid-related imaging abnormalities (ARIA): radiological, biological and clinical characteristics. Brain, 146(11), 4414-4424.
[25]. Zhang, Z., Yang, X., Song, Y. Q., and Tu, J. (2021) Autophagy in Alzheimer's disease pathogenesis: Therapeutic potential and future perspectives. Ageing Research Reviews, 72, 101464.
[26]. Qin, Q., Teng, Z., Liu, C., Li, Q., Yin, Y., and Tang, Y. (2021) TREM2, microglia, and Alzheimer’s disease. Mechanisms of Ageing and Development, 195, 111438.
[27]. Zhao, X., Hu, Q., Wang, X. et al. (2024) Dual-target inhibitors based on acetylcholinesterase: Novel agents for Alzheimer's disease. European Journal of Medicinal Chemistry, 279, 116810.
[28]. Vemula, P., Schoch, K. M., and Miller, T. M. (2023) Evaluating the efficacy of purchased antisense oligonucleotides to reduce mouse and human tau in vivo. Frontiers in Molecular Neuroscience, 16, 1320182.
[29]. Cao, Z., Kong, F., Ding, J., Chen, C., He, F., and Deng, W. (2024) Promoting Alzheimer’s disease research and therapy with stem cell technology. Stem Cell Research & Therapy, 15(1), 136.
[30]. Wang, J., Jin, C., Zhou, J., Zhou, R., Tian, M., Lee, H. J., and Zhang, H. (2022) PET molecular imaging for pathophysiological visualization in Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging, 50(3), 765-783.