







1. Introduction
Tissues of multicellular species are composed of various cell subpopulations. Their physiological processes and functions are closely related to spatial distribution and cell interactions. A deeper understanding of tissue structure and heterogeneity, along with insights into intercellular communication and the biological significance of microenvironments, can provide crucial support for advancements in life sciences.
The brain, as one of the most complex organs, has regional delineation that is particularly important in neuroscience research. This delineation not only aids in understanding the different functional areas of the brain and their interrelationships but also promotes the development of cognitive science. By clarifying the functions of each region, scientists can more effectively reveal the mechanisms of brain operation. Furthermore, partitioning brain regions is vital for neuroscience and medicine, as it helps clinicians accurately locate areas of brain injury, allowing for personalized treatment plans.
In the past decade, single-cell RNA sequencing (scRNA-seq) has become an essential tool in biomedical research, particularly in fields such as developmental biology, cancer, immunology, and neuroscience. Recently, spatial transcriptomics has rapidly advanced, enabling quantitative analysis of gene expression within the spatial context of tissues. Compared to traditional transcriptomics, spatial transcriptomics not only detects gene expression levels within cells but also reveals the spatial locations of genes within tissues, cell composition, and intercellular interactions. This technology allows for the acquisition of transcriptomic data from various locations within a complete tissue sample.
Currently, spatial transcriptomics can be categorized into two major types: in situ capture and sequencing technologies, and imaging-based spatial transcriptomics [1]. These technologies have been widely applied in neurobiology, developmental biology, and tumor biology, enhancing our understanding of complex biological systems. In situ transcript capture and sequencing techniques utilize DNA-tagged primers with spatial location information to capture and label transcripts, thereby constructing spatial maps of multi-omics gene expression. These techniques primarily target tissue samples and provide untargeted detection, covering genes across the entire transcriptome. They are suitable for exploring unknown biomarkers and emerging cell types. Their advantage lies in offering a comprehensive view of gene expression within tissues; however, the lack of specificity may result in insufficient resolution for certain genes. In contrast, imaging-based spatial transcriptomics techniques, such as in situ sequencing with lock-type probes and hybridization detection with complementary fluorescent probes, allow for targeted detection using individual cells as samples. This enables researchers to focus on the expression of specific genes and their spatial distribution, thereby revealing intercellular interactions and cellular heterogeneity within tissues. However, the throughput of these techniques often does not cover the entire transcriptome, which limits their application for certain biological questions.
In this study, we utilized 10X Visium sequencing data from the mouse brain. By employing artificial priors and the SpaceFlow method [2], we identified the layered structure of the mouse hindbrain. Subsequently, we performed differential gene expression and Gene Ontology (GO) analyses on the MO::L1 and MO::L2/3 regions to uncover the key functions and biological processes associated with these areas.
2. Transcriptomics
Transcriptome sequencing technology is a crucial tool for studying gene expression. It involves a comprehensive analysis of all transcripts within a cell or tissue, revealing the functions and regulatory mechanisms of genes in various biological processes. As technology continues to advance, transcriptome sequencing has developed multiple methods to accommodate different research needs.
Bulk RNA-seq is one of the earliest widely applied transcriptome technologies. By extracting and sequencing RNA from entire samples, it provides the average gene expression levels across all cells within a sample. This technique offers high throughput and relatively low costs, enabling large-scale biological research and allowing for the analysis of complex tissues or organisms. Bulk RNA-seq is particularly useful for identifying global changes in gene expression, providing insights into developmental processes, disease states, and responses to treatments. However, the primary drawback of bulk RNA-seq is its inability to reveal cellular heterogeneity within tissues. Since the expression data from all cells are mixed together, it leads to a loss of information regarding intercellular interactions and the microenvironment. This averaging effect can mask the expression profiles of rare cell types or subpopulations, which may play crucial roles in biological functions and disease mechanisms. Consequently, bulk RNA-seq limits our understanding of complex biological systems, as it does not capture the nuances of cellular diversity and dynamic behavior within tissues. In addition, bulk RNA-seq can complicate the interpretation of results, especially when studying heterogeneous tissues where different cell types may have opposing roles or expression patterns. Researchers may miss critical insights into cellular interactions, signaling pathways, and microenvironmental influences.
To address these limitations, scRNA-seq has been introduced. This technology allows researchers to analyze gene expression at the single-cell level, uncovering cellular heterogeneity within complex tissues. The advent of scRNA-seq has enabled scientists to identify various cell types and states, facilitating a deeper exploration of unique cellular roles in biological processes. By employing this method, researchers can obtain detailed expression profiles for individual cells, enhancing our understanding of how cells respond to environmental changes, participate in developmental processes, and function in disease states. The strength of scRNA-seq lies in its ability to capture information about rare cell types within a tissue, which are often overlooked in traditional bulk RNA sequencing. This technique also allows for stratified analysis of cell populations, identifying specific subpopulations and their functions. For instance, in cancer research, scRNA-seq can reveal the interactions and contributions of different cell types within the tumor microenvironment, providing new insights for targeted therapies. However, despite its significant advantages in elucidating cellular diversity, scRNA-seq faces several challenges. The processes of cell isolation and handling can lead to the loss of intercellular interactions and microenvironmental information. Furthermore, scRNA-seq is typically performed on tissue sections, which limits the spatial localization of data, thereby hindering our ability to accurately reflect cellular behavior within its natural environment. These factors impede a comprehensive understanding of cellular dynamics, particularly in complex biological systems. Additionally, the data generated by scRNA-seq is complex and requires extensive bioinformatics analysis. Researchers must develop advanced algorithms and tools to process and interpret these high-dimensional data sets to extract meaningful biological information. Nevertheless, scRNA-seq remains a crucial technology in modern biomedical research, particularly for understanding developmental mechanisms, disease processes, and intercellular interactions.
To further overcome these challenges, spatial transcriptomics emerged. This approach aims to combine gene expression analysis with spatial information from tissue samples. Spatial transcriptomics utilizes methods such as in situ hybridization and sequencing to capture the spatial distribution of transcripts, revealing intercellular interactions and their functions in specific microenvironments. A significant advantage of this method is its ability to obtain detailed gene expression information while preserving tissue architecture, allowing researchers to analyze complex cellular dynamics in their natural settings.
In summary, the evolution of transcriptome sequencing technology has progressed from bulk RNA-seq to single-cell transcriptomics, and finally to spatial transcriptomics. Each technique has its own advantages and disadvantages. Bulk RNA-seq excels in sample processing and cost but lacks insights into cellular diversity and spatial distribution. Single-cell transcriptomics performs well in elucidating cellular heterogeneity but may lose spatial information. In contrast, spatial transcriptomics combines the strengths of both approaches, providing in-depth understanding of cellular interactions and tissue structure.
3. Datasets and Methods
In our study, we utilized mouse posterior brain samples that were processed using the 10X Genomics Visium sequencing technology [3]. This technology enables high-resolution spatial transcriptomics, allowing researchers to capture gene expression profiles while preserving the spatial context of the tissue
Our analysis focused on the MO::L1 and MO::L2/3 regions of the mouse posterior brain. To delineate spatial regions, we first employed SpaceFlow, a deep learning-based analytical method specifically designed for spatial transcriptomics data. SpaceFlow aims to identify spatial regions within tissues and uncover spatially correlated molecular features. This method integrates gene expression data, spatial location information, and potentially histological images to capture molecular characteristics across different areas of the tissue. By combining prior knowledge with automated clustering algorithms, SpaceFlow optimizes the delineation of spatial regions. This approach enhances our understanding of tissue heterogeneity and reveals functionally distinct areas. After data input, we performed standard preprocessing steps. Subsequently, we refined the manually annotated spatial regions using the results from SpaceFlow. This refinement process is significant as it combines prior knowledge with molecular features, leading to a more accurate delineation of spatial regions. By integrating both aspects, we achieve a clearer understanding of the spatial organization and functional diversity within the tissue.
We employed DEsingle as our differential analysis method [4]. DEsingle is an R package specifically designed for the analysis of differential expression in scRNA-seq data. This tool aims to define and detect three types of differentially expressed genes between two groups of single cells: different expression state genes, differential expression abundance genes, and general differential expression genes. DEsingle utilizes a zero-inflated negative binomial model, which effectively estimates the proportion of true zeros and missing zeros, thereby accurately defining and detecting these three categories of differential genes. The advantage of DEsingle lies in its ability to outperform existing methods for scRNA-seq differential expression analysis, revealing various types of differential genes with distinct biological functions. This capability enables researchers to gain deeper insights into functional differences between cells, providing crucial leads for subsequent biological studies.
In our research, we focused on the differential gene analysis of the MO::L1 and MO::L2/3 regions. To select differential genes, we set the thresholds: a fold change of at least 2 and a p-value of less than 0.05. This criterion ensures that the selected differential genes are statistically significant and biologically relevant. Through this approach, we aim to identify genes with significant functional differences in these two regions, thereby supporting a better understanding of their biological characteristics.
4. Results
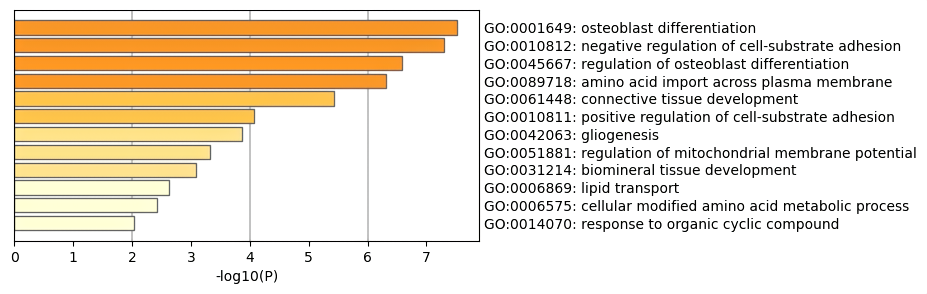
Figure 1. GO analysis results.
In this study, we conducted Gene Ontology (GO) analysis on the selected differentially expressed genes using Metascape [5]. Metascape is a powerful online tool that assists researchers in gene functional annotation and enrichment analysis. With this tool, we were able to identify the enrichment of differentially expressed genes across various biological processes, cellular components, and molecular functions.
During the analysis, we input the list of selected differentially expressed genes into Metascape, which automatically generated relevant GO terms and calculated the enrichment significance for each term. Among these terms (see Figure 1), we observed that the main entries were primarily related to common developmental processes. For instance, GO:0001649 refers to the osteoblast differentiation process, which is crucial for bone tissue formation and mineralization. This process plays a significant role in bone generation and skeletal development and is also important in fracture healing and bone remodeling. Abnormal osteoblast differentiation can lead to skeletal diseases such as osteoporosis and osteogenesis imperfecta. Additionally, GO:0045667 pertains to the regulation of osteoblast differentiation, controlling the rate and extent of osteoblast generation. This regulatory process is vital for bone formation and maintaining bone density, and any disruption can result in metabolic disorders such as osteoporosis or osteosclerosis. Another relevant term, GO:0061448, relates to the development of connective tissue, which involves the formation of supporting tissues such as bone, tendons, and ligaments. Connective tissue provides structural support to organs and tissues and participates in immune responses and wound healing. Abnormal development of connective tissue can lead to disorders such as scleroderma and rheumatoid arthritis. Interestingly, we found that GO:0042063 is associated with the process of generating glial cells, including the production of glial progenitor cells and their differentiation into mature glial cells.
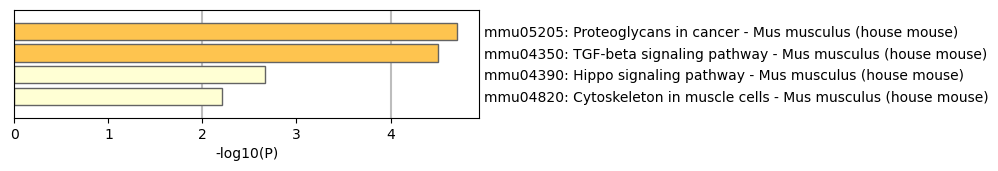
Figure 2. KEGG pathway.
Furthermore, through the study of differentially expressed genes in the brain, we identified several key KEGG pathways (see the Figure 2), including the proteoglycans in cancer (mmu05205), TGF-beta signaling pathway (mmu04350), Hippo signaling pathway (mmu04390), and cytoskeleton in muscle cells (mmu04820). Among these, the TGF-beta signaling pathway is crucial for neurodevelopment and injury repair, but its overactivation may lead to neurological dysfunction. The Hippo signaling pathway regulates cell proliferation and growth, influencing the fate of neural stem cells and is associated with the occurrence of brain tumors. Finally, the cytoskeleton plays a critical role in maintaining neuronal morphology and function, affecting synapse formation and related cognitive functions. The interactions among these pathways provide important insights into understanding the health and disease states of the brain.
5. Conclusion
In this study, we analyzed spatial transcriptomic data from the MO::L1 and MO::L2/3 regions of the mouse posterior brain. Through this analysis, we successfully identified differentially expressed genes (DEGs) and revealed their significant roles in developmental processes, cellular regulation, and signaling pathways via Gene Ontology (GO) and KEGG pathway analysis. Specifically, we identified key signaling pathways, such as the TGF-beta signaling pathway (mmu04350), which plays a vital role in neurodevelopment and injury repair; the Hippo signaling pathway (mmu04390), which regulates cell proliferation and neural stem cell fate; and the cytoskeleton in muscle cells (mmu04820), which is crucial for maintaining neuronal morphology and function. These findings provide important insights into the functional characteristics and molecular mechanisms of brain regions.
However, this study does have some limitations and areas for improvement. Although we employed the SpaceFlow method for spatial region delineation, the accuracy of the algorithm may be affected by data quality and processing steps in certain cases. To address this issue, future research could explore other spatial transcriptomic analysis tools, such as Seurat [6], which may offer greater precision and flexibility. Additionally, while the application of DEsingle in differential gene analysis was effective, further validation of its stability and reliability under different experimental conditions is necessary. We recommend conducting repeat experiments across multiple experimental setups to evaluate the performance of DEsingle and to compare it with other differential expression analysis methods to determine best practices.
This study primarily focused on mouse models, and future work should consider different species and pathological conditions to enhance the generalizability of the results. Such cross-species comparisons can help identify conserved biological mechanisms and provide a more reliable foundation for studying human diseases. Moreover, increasing the sample size can significantly enhance the statistical power of the analysis, reducing the likelihood of false negatives and thereby improving the credibility and reproducibility of the research. Integrating spatial transcriptomic data with other omics data, such as proteomics and metabolomics, will aid in comprehensively understanding intercellular interactions and overall biological processes. This multi-omics integration approach can reveal potential regulatory networks and biomarkers, thereby providing new targets and strategies for disease diagnosis and treatment. By merging data across different layers, we can construct a more comprehensive biological network, facilitating deeper analysis of cellular functions and signaling pathways.
With the advancement of deep learning models, future research can leverage these advanced technologies to more accurately identify complex cell types and states, thereby elucidating intercellular interactions [3, 7, 8]. The powerful computational capabilities of deep learning allow it to handle large datasets and discover subtle patterns those traditional methods may miss, which is especially important in neuroscience research. Furthermore, advances in 3D slice transcriptomics enable us to achieve a three-dimensional reconstruction of gene expression within tissues, leading to a more comprehensive understanding of the relationship between tissue structure and function, particularly during development and pathology. This technology not only improves spatial resolution but also reveals the distribution and interactions of cells in three-dimensional space.
Currently, this research primarily focuses on static data analysis. Future studies could consider dynamic monitoring across different developmental stages or pathological conditions. This approach will help us understand the spatiotemporal changes in gene expression and their roles in neurodevelopment and disease. By capturing the state changes of cells at specific time points, we can reveal their roles in developmental and disease processes. This combination of 3D transcriptomics and deep learning methods can provide a more detailed and dynamic biological perspective, advancing our understanding of the complexities of the nervous system. Moreover, extending these technologies to other tissues and systems will provide a broader biological perspective. Differences in cell types and functions across various tissues may influence gene expression patterns and their regulatory mechanisms. Through cross-tissue studies, we can better understand the interactions and regulatory networks of different biological systems. By pursuing these comprehensive research directions, we aim to achieve deeper breakthroughs in the biomedical field, particularly in understanding and treating neuro-related diseases, thus promoting the development of precision medicine.
By addressing these improvement directions, researchers will be able to explore the functional characteristics and molecular mechanisms of the nervous system more comprehensively, laying a solid foundation for future biomedical research.
References
[1]. Y. Lu, Q. M. Chen, and L. An, “SPADE: spatial deconvolution for domain specific cell-type estimation,” Commun Biol, vol. 7, no. 1, pp. 1–12, Apr. 2024, doi: 10.1038/s42003-024-06172-y.
[2]. H. Ren, B. L. Walker, Z. Cang, and Q. Nie, “Identifying multicellular spatiotemporal organization of cells with SpaceFlow,” Nat Commun, vol. 13, no. 1, p. 4076, Jul. 2022, doi: 10.1038/s41467-022-31739-w.
[3]. J. Hu et al., “SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network,” Nat Methods, vol. 18, no. 11, pp. 1342–1351, Nov. 2021, doi: 10.1038/s41592-021-01255-8.
[4]. Z. Miao, K. Deng, X. Wang, and X. Zhang, “DEsingle for detecting three types of differential expression in single-cell RNA-seq data,” Bioinformatics, vol. 34, no. 18, pp. 3223–3224, Sep. 2018, doi: 10.1093/bioinformatics/bty332.
[5]. Y. Zhou et al., “Metascape provides a biologist-oriented resource for the analysis of systems-level datasets,” Nat Commun, vol. 10, no. 1, p. 1523, Apr. 2019, doi: 10.1038/s41467-019-09234-6.
[6]. R. Satija, J. A. Farrell, D. Gennert, A. F. Schier, and A. Regev, “Spatial reconstruction of single-cell gene expression data,” Nat Biotechnol, vol. 33, no. 5, pp. 495–502, May 2015, doi: 10.1038/nbt.3192.
[7]. T. Biancalani et al., “Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram,” Nat Methods, vol. 18, no. 11, Art. no. 11, Nov. 2021, doi: 10.1038/s41592-021-01264-7.
[8]. R. Lopez et al., “DestVI identifies continuums of cell types in spatial transcriptomics data,” Nat Biotechnol, vol. 40, no. 9, Art. no. 9, Sep. 2022, doi: 10.1038/s41587-022-01272-8.
Cite this article
Piao,M. (2024). Deciphering Biological Function Difference Between MO::L1 and MO::L2/3 in Mouse Brain from Spatial Transcriptomics Data. Theoretical and Natural Science,67,159-164.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Y. Lu, Q. M. Chen, and L. An, “SPADE: spatial deconvolution for domain specific cell-type estimation,” Commun Biol, vol. 7, no. 1, pp. 1–12, Apr. 2024, doi: 10.1038/s42003-024-06172-y.
[2]. H. Ren, B. L. Walker, Z. Cang, and Q. Nie, “Identifying multicellular spatiotemporal organization of cells with SpaceFlow,” Nat Commun, vol. 13, no. 1, p. 4076, Jul. 2022, doi: 10.1038/s41467-022-31739-w.
[3]. J. Hu et al., “SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network,” Nat Methods, vol. 18, no. 11, pp. 1342–1351, Nov. 2021, doi: 10.1038/s41592-021-01255-8.
[4]. Z. Miao, K. Deng, X. Wang, and X. Zhang, “DEsingle for detecting three types of differential expression in single-cell RNA-seq data,” Bioinformatics, vol. 34, no. 18, pp. 3223–3224, Sep. 2018, doi: 10.1093/bioinformatics/bty332.
[5]. Y. Zhou et al., “Metascape provides a biologist-oriented resource for the analysis of systems-level datasets,” Nat Commun, vol. 10, no. 1, p. 1523, Apr. 2019, doi: 10.1038/s41467-019-09234-6.
[6]. R. Satija, J. A. Farrell, D. Gennert, A. F. Schier, and A. Regev, “Spatial reconstruction of single-cell gene expression data,” Nat Biotechnol, vol. 33, no. 5, pp. 495–502, May 2015, doi: 10.1038/nbt.3192.
[7]. T. Biancalani et al., “Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram,” Nat Methods, vol. 18, no. 11, Art. no. 11, Nov. 2021, doi: 10.1038/s41592-021-01264-7.
[8]. R. Lopez et al., “DestVI identifies continuums of cell types in spatial transcriptomics data,” Nat Biotechnol, vol. 40, no. 9, Art. no. 9, Sep. 2022, doi: 10.1038/s41587-022-01272-8.