







1. Introduction
Cancer is the second leading cause of death worldwide [1], with an approximated 20 million new cases of cancer in the year 2022 and nearly 9.7 million fatalities attributable to cancer [2]. Notwithstanding considerable progress in therapeutic approaches—including surgery, chemotherapy, radiotherapy, targeted therapy, and immunotherapy—many cancers develop resistance, relapse, or remain unresponsive to current interventions. A pivotal challenge in cancer treatment is the immunosuppressive nature of the tumor microenvironment (TME), which fosters tumor immune evasion and limits the efficacy of immunotherapies [3].
Macrophage polarization plays a dual role in tumor development, significantly influencing cancer progression and therapeutic outcomes. M1 macrophages, also known as classically activated macrophages, inhibit tumor growth by releasing proinflammatory factor (such as TNFα, IL-6, IL-8, and chemokines, such as CXCL10, which enhance anti-tumor immunity and directly kill cancer cells. Conversely, M2 macrophages, or alternatively activated macrophages, promote tumorigenesis by secreting anti-inflammatory factors (such as IL-10, TGF-β, and VEGFA.), which facilitate angiogenesis, immune evasion, and metastasis [4-7]. The TME plays a crucial role in shaping macrophage polarization, often favoring the M2 phenotype through cytokines produced by tumor cells and stromal components [8]. This shift creates an immunosuppressive milieu that supports tumor survival and progression. Additionally, hypoxia and extracellular matrix remodeling further reinforce M2 polarization, establishing a feedback loop that sustains tumor growth [9]. Understanding the mechanisms behind macrophage polarization offers potential therapeutic strategies, such as reprogramming M2 macrophages to the M1 phenotype or blocking M2-associated signaling pathways, to enhance anti-tumor immunity and improve cancer treatment efficacy.
A critical challenge is the role of innate immune signaling dysregulation in cancer. TANK binding kinase 1 (TBK1), as a key kinase in specific signaling pathways, often exhibits abnormal activation in various tumors. Its excessive activation can promote the massive production of type I interferons (IFNs) and various pro-inflammatory cytokines; However, these factors do not enhance anti-tumor immunity, but rather promote tumor growth and maintenance in a contradictory way [8]. Through activation of the TBK1-IRF3 axis, TRIM21 plays a regulatory role in TBK1 signaling [9]. Furthermore, dysregulation of Tripartite motif containing protein 21 (TRIM21) has been linked to autoimmune diseases and cancer but its role in shaping the TME remains incompletely understood [9].
Moreover, inhibitor of DNA binding 3 (ID3), a transcriptional regulator, has been implicated in immune cell exhaustion and tumor plasticity [10]. High ID3 expression correlates with poor prognosis in multiple cancers. Despite its emerging significance, the interplay between ID3 and inflammatory signaling pathways within the TME has not been thoroughly investigated [9].
Current therapeutic strategies have shown limited success due to compensatory signaling pathways and systemic side effects. Although immune checkpoint blockade has transformed cancer therapy, only a subset of patients achieves durable responses, underscoring the need for novel approaches that target alternative immune evasion mechanisms. Therefore, identifying the possible mechanisms among TRIM21/ TBK1/ ID3 could provide a more effective means of reversing immunosuppression and enhancing therapeutic response.
In the context of macrophage polarization, several interconnected signaling routes converge on TBK1 as a central node. The TBK1–IRF4/IRF5 axis modulates the balance between immunosuppressive M2 and proinflammatory M1 states, with TBK1 activity favoring IRF4-driven M2 differentiation, whereas its inhibition shifts macrophages toward IRF5-dependent M1 activation. ID3, through its interaction with E proteins such as E2A, can reshape transcriptional programs that influence this balance, potentially limiting the expression of genes supportive of M2 phenotypes. ELK1, a transcription factor whose function depends on its cofactors, may cooperate with ID3 and E2A in ways that alter promoter accessibility and gene regulation relevant to TBK1 expression. Together, these pathways form a mechanistic framework in which modulation of TBK1 activity can reprogram macrophage phenotype within the tumor microenvironment.
ID3 is a transcription factor belonging to the helix loop helix (HLH) family, which does not contain typical DNA binding domains in its structure, but can heterodimerize with E proteins such as E2A. This binding sequesters E2A, preventing it from binding to E-box motifs in promoter regions, including those of immune regulatory genes such as TBK1. This suppression of E2A activity rewires transcriptional programs relevant to macrophage differentiation [14].
ELK1 is a member of the ETS-domain transcription factor family. It typically binds ETS motifs and functions in gene activation or repression depending on its cofactors. Evidence shows that ID3 can upregulate and interact with ELK1, and this interaction can drive apoptotic or immune signaling pathways. In macrophages, ID3 may recruit ELK1 into the ID3–E2A complex or alter its genomic binding profile through competitive binding dynamics [14].
Drawing from tumor and macrophage models, researchers hypothesize that the ID3–E2A–ELK1 complex either occupies the TBK1 promoter or modifies local chromatin, repressing transcription. ELK1, when associated with ID3 and E2A, may switch from an activator to a transcriptional repressor, possibly by recruiting histone deacetylases (HDACs) or other co-repressors [8,14].
TBK1 positively promotes macrophage polarization towards M2 phenotype by regulating the activation of IRF4 and inhibition of IRF5. Its inhibition leads to a shift toward M1 polarization, marked by increased IRF5 activity and elevated expression of proinflammatory markers like IL-1β, TNF-α, and CD86, while suppressing M2 markers like ARG1 [15].
Thus, the ID3–E2A–ELK1 complex serves as a critical upstream switch that represses TBK1 transcription, disinhibits IRF5, and promotes M1-type macrophage polarization. This mechanistic axis may represent a therapeutic target for enhancing antitumor immunity [16,17].
Our strategy draws upon insights from three prior studies, each of which explored distinct orthologous approaches in the realms of pharmacology and immunotherapy.
2. Literature review
Yuanyuan Li et al. [10] explores TBK1 in macrophage polarization in the tumor microenvironment, especially glioblastoma multiforme (GBM). In GBM, tumor-associated macrophages (TAMs) often develop an immunosuppressive M2 phenotype under macrophage colony-stimulating factor (M-CSF). TBK1 is highly expressed and phosphorylated in glioma-infiltrating CD11b⁺ myeloid cells, particularly M-CSF-stimulated macrophages. Conditional TBK1 knockout in mice shifts M-CSF-induced bone marrow-derived macrophages to a proinflammatory M1-like phenotype, with upregulated IL-1β, TNF-α, CD86 and downregulated Argl. Mechanistically, TBK1 inhibition/reduction decreases IRF4 but increases IRF5 activation. IRF5 deletion reverses TBK1 inhibition effects, confirming TBK1 acts via the IRF5/IRF4 axis. TBK1 activation is independent of the canonical IRF3 pathway. These findings identify TBK1’s role in maintaining TAMs’ M2 phenotype, suggesting TBK1 inhibition may reprogram macrophages to suppress GBM.
Zihou Deng et al. [11] reveals that the transcription factor ID3 enhances the anti-tumor capacity of Kupffer cells (KCs) by regulating the equilibrium between inhibitory (SIRPA) and activating (dectin-1) receptors. Using genetic models, RNA-seq, CUT&RUN, and in vitro/in vivo assays, the authors showed that ID3 represses SIRPA by inhibiting E2A and ELK1 binding to its promoter/enhancer, enhancing phagocytosis of live tumor cells. ID3-deficient KCs exhibited impaired tumor clearance and reduced lymphoid effector recruitment (NK and CD8+T cells). SIRPA blockade or CD47 deletion rescued these defects. Ectopic ID3 expression in mouse BMDMs and human iPSC-derived macrophages conferred similar anti-tumor activity, suggesting therapeutic potential. The findings highlight ID3 as a key regulator of macrophage-mediated tumor immunity, orchestrating phagocytosis, and immune niche formation.
Ruihong Zhang et al. [12] explores the role of TRIM21-mediated ubiquitination of Sohlh2, a basic helix-loop-helix transcription factor, in suppressing M2 macrophage polarization and triple-negative breast cancer (TNBC) development. Results indicate that Sohlh2 is upregulated in M2 macrophages and facilitates cholesterol efflux via activation of the LXRα/ABCA1/ABCG1 signaling axis, leading to disrupted lipid homeostasis and enhanced M2 polarization. These effects consequently promote the proliferation, migration, and lung metastasis of TNBC cells. At the molecular level, Sohlh2 directly interacts with the LXRα promoter and upregulates its transcription. The E3 ubiquitin ligase TRIM21 counteracts these processes by ubiquitinating Sohlh2 and targeting it for proteasomal degradation. Overexpression of TRIM21 was shown to attenuate the pro-tumorigenic and polarization-promoting functions of Sohlh2. Analysis of clinical specimens further revealed a positive correlation between Sohlh2 expression and levels of the M2 marker CD163 as well as LXRα. These results highlight the importance of the TRIM21/Sohlh2/LXRα axis in modulating macrophage polarization and TNBC progression, suggesting its potential as a therapeutic target for TNBC treatment.
In summary, we present a proposal for studying the mechanisms and interactions among TRIM21/TBK1/ID3 to provide new targets for cancer therapy in the future.
3. Overarching hypothesis
We hypothesize that ID3, as an inhibitor of DNA binding with multiple binding effects, forms an ID3-E2A/ELK1 complex by binding to E proteins, acts on the promoter region of TBK1, thereby inhibiting the expression of TBK1, and achieving the goal of promoting the differentiation of macrophages in the microenvironment into the M1 type, as shown in Figure 1.
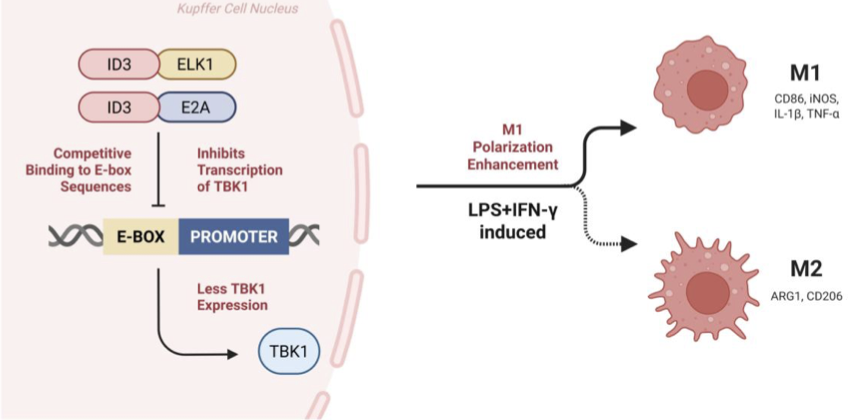
4. Working model
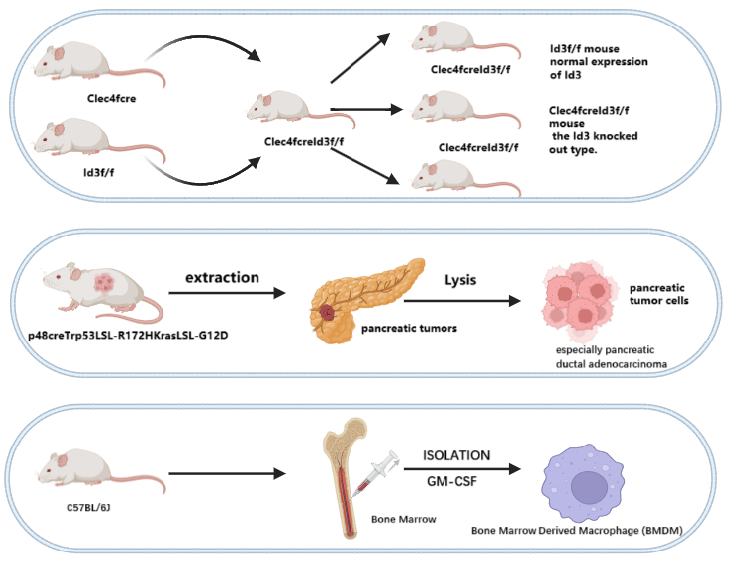
The methods used to produce our models are partially shown in Figure 2.
4.1. Mouse models
We propose to establish Id3f/f [18]and Clec4fcreId3f/fmouse based on C57BL/6J [11]. Clec4fcreId3f/fis the offspring of a cross between Id3f/f and Clec4fcre [19]. Id3f/fmouse has normal expression of Id3, while Clec4fcreId3f/f mouse is the Id3-knocked out type.
p48creTrp53LSL-R172HKrasLSL-G12D [20]is used for tumor cell extraction.
4.2. Cell models
Kupffer cells (KC) from Id3f/fand Clec4fcreId3f/fmouse littermates
Bone Marrow Derived Macrophage (BMDM) mouse primary cells extracted from femur, tibia and iliac bones of C57BL/6J mice.
Human Induced Pluripotent Stem Cells – Macrophages (hiPSC – Macs).
KPC-1 and KPC-2 cell lines obtained from pancreas tissue from p48creTrp53LSL-R172HKrasLSL-G12D mice. Labeled with Cas-green for observation.
5. Visual depiction
6. Specific aims
1) To verify that ID3-E2A/ELK1 binds to the TBK1 promoter and inhibits TBK1 expression.
2) To verify that inhibition of TBK1 expression by ID3 enhances macrophage phagocytosis.
3) To verify that inhibition of TBK1 expression by ID3 promotes macrophage M1 polarization.
7. Approach
To verify that ID3-E2A/ELK1 binds to the TBK1 promoter and inhibits TBK1 expression, deep learning analysis will be first utilized to identify E-Box-binding sites/ ELK1 binding sites in the TBK1 promoter region upstream of mouse KCs and BMDMs.
To clearly demonstrate that E2A/ELK1 can bind to the promoter region of TBK1, first, download the approximate sequence of the TBK1 promoter region (2 kb upstream and downstream of the transcription start site, TSS) from NCBI. Then, combine it with ATAC-seq/H3K27ac data of KCs/BMDMs to screen regions near the TBK1 promoter that have ATAC-seq peaks and high H3K27ac signals, thereby further refining the candidate regulatory regions. Establish a positive dataset from the localized locus regions based on an H3K27ac tag count (≥32), and the negative dataset consists of random sequences that are GC-matched to the positive dataset. Next, use a pre-trained DeepSEA model [16], fine-tune it with the dataset, and then combine it with DeepLIFT analysis to calculate nucleotide importance scores, so as to narrow down the potential range. Finally, combine the PWM matrices of E2A (MA0522.1) and ELK1 (MA0028.2) downloaded from the JASPAR database [21] with the importance scores analyzed by DeepLIFT to obtain the final specific regulatory positions.
After confirming the binding site, a CUT& RUN assay will be performed on E2A with ELK1 in KCs from Clec4fcreId3f/fmice and Id3f/flittermates (n = 3 per group) to see the extent of E2A/ELK1 binding to the TBK1 promoter region. To get E2A/ELK1 knocked-out BMDMs and KCs, lentiviral supernatant carrying lenti-control, shE2A, shELK1will be added to cell culture medium, then select the cells with 1ug ml-1 puromycin and analyze the transduced cells by FACS. Flow cytometry will be employed to analyze TBK1 expression in two distinct cell populations: 1) Kupffer cells (KCs) isolated from Id3+/+ mice and their Id3-/- littermates, with these KCs expressing scramble, E2a, or ELK1 short hairpin RNAs (shRNAs); and 2) mouse bone marrow-derived macrophages (BMDMs) that express either lenti-control or the indicated shRNAs. The sample size for each experimental group is set at n = 3.
To verify that Id3 enhances macrophage phagocytosis by inhibiting TBK1 expression, BMDMs or hiPSC-Macs will be divided into two groups: lenti-ctrl group (blank control group): Cells are transduced with a control lentivirus that does not carry the Id3 overexpression construct and lenti-Id3 group: Cells are transduced with a lentivirus engineered to overexpress Id3.Subsequently, RT-qPCR and flow cytometry will be performed to detect whether TBK1 content decreases in cells after lentiviral transduction-induced Id3 overexpression.
For the in vitro engulfment assay, an ex vivo 3D co-culture will be conducted overnight using lentivirus-transduced cells and Cas-Green-KPC-1-mtdT cells, with Matrigel serving as the culture matrix. To visualize the interaction between Kupffer cells (KCs) and KPC-1-mtdT tumor cells, time-lapse imaging will be performed using a confocal laser-scanning microscope. Subsequently, the percentage of phagocytic KCs will be determined by calculating the average of the proportions of cells that have engulfed live tumor cells.
In vivo experiments. C57BL/6 mouse derived KPC-1 cells will be injected into 6-week-old syngeneic Clec4fcreId3f/f mice to construct the model, while a certain number of BMDM overexpressing Id3 in a range (103 - 107) is injected into BMDM versus control BMDM, and tumor growth and weight of the control mice is observed using living imaging, and the tumors are taken out to assess the tumor status after two weeks of incubation. The tumors will be removed after two weeks of incubation to assess the tumor condition.
To verify that ID3-mediated inhibition of TBK1 expression promotes M1 polarization of macrophages, the mRNA and protein expression levels of M1 macrophage-associated genes (TNFα, IL-1β, IL-6, IL-8, iNOS), and M2 macrophage-associated genes (e.g., TGFβ, IL-10, CD163, CD206, Arg1, Fizz1, YM1) will be measured in ID3 overexpressing cells(lenti-ID3) and control groups. This analysis will be performed using quantitative polymerase chain reaction (qPCR) and Western blotting, following treatment of the cells with lipopolysaccharide (LPS) plus interferon-γ (IFNγ).
8. Anticipated results
8.1. CUT&RUN assay (E2A/ELK1 binding)
Expected Outcome:
The data from Clec4fcreId3f/f mice indicate that the binding force of E2A/ELK1 to the TBK1 promoter is very strong. (see Fig.3)
In the Id3f/f sibling group, due to the disruption of the ID3-E2A/ELK1 complex. A reduction or complete loss of E2A/ELK1 binding at the TBK1 promoter was observed. (see Fig.3)
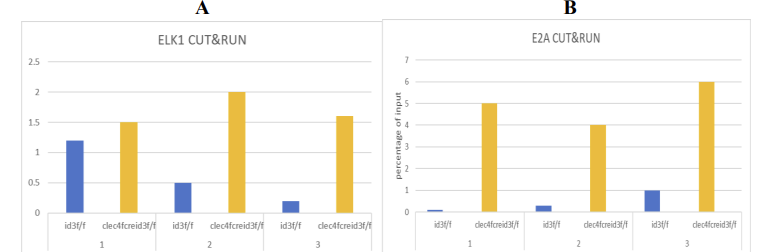
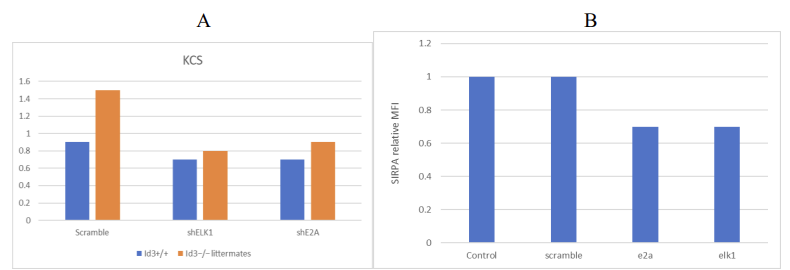
8.2. Compared to the scramble shRNA, the protein level of E2A/ELK1 in BMDM cells is decreased
Expected Outcome:
Scramble shRNA: Normal TBK1 expression. (Fig.4A)
shE2A/shELK1: Due to the inhibition of the expression of the complex, the overexpression of TBK1 was caused. (Fig.4)
8.3. ID3 overexpression in BMDMs/hiPSC-Macs
Expected Outcome:
The TBK1 in the lenti-mID3 group was at a basal level.
lenti-ctrl: The mRNA/protein levels of TBK1 were decreased.
8.4. In vitro engulfment assay (KPC-1 tumor cells)
Expected Outcome:
Lenti-mID3: The proportion of macrophages that engulf Cas-green tumor cells is higher. Lenti-ctrl: The engulfment rate is lower. (see Fig.5)
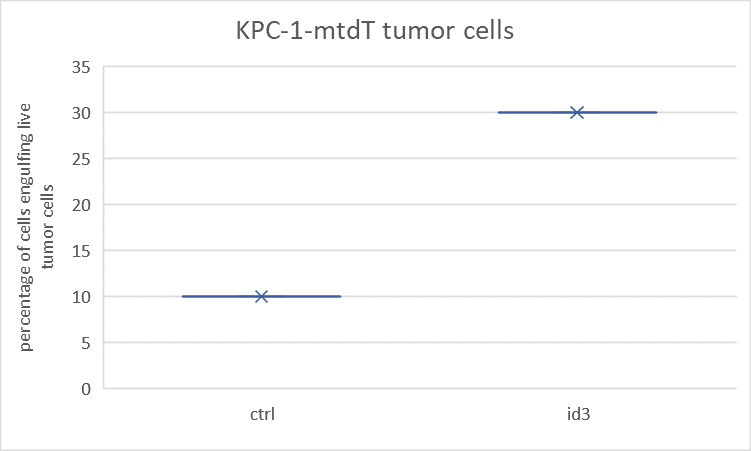
8.5. In vivo tumor model (Clec4fcreid3f/f Mice)
Expected Outcome:
Fig.6 shows the survival rates of the no BMDM group and the BMDM lenti-ctrl group dropped linearly to 0 two weeks later.
The BMDM lentimid3 group began to die after three weeks and remained at 50% level thereafter. (see Fig.6)
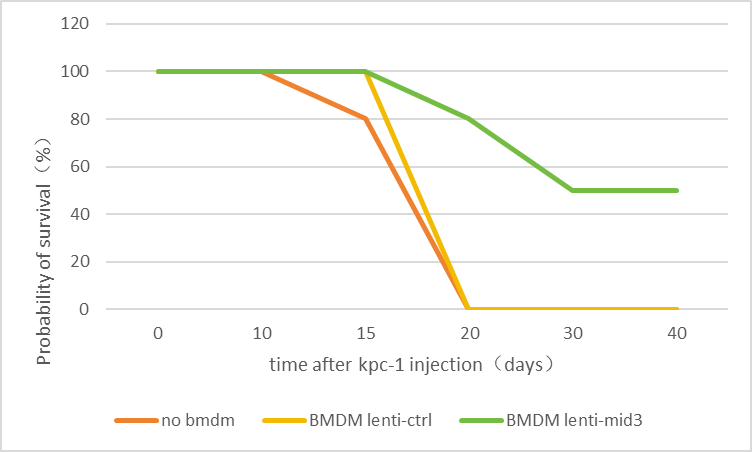
8.6. Verify the inhibitory effect of ID3 on the expression of TBK1 and its impact on macrophage polarization
Expected Outcome:
The signal levels of lenti-id3l:m1 type macrophages were much higher than those of lenti-ctrl group. (Fig.7A) On the other hand, the levels of lenti-id3:m2 type macrophages were much lower than those of lenti-ctrl group. (Fig.7B)
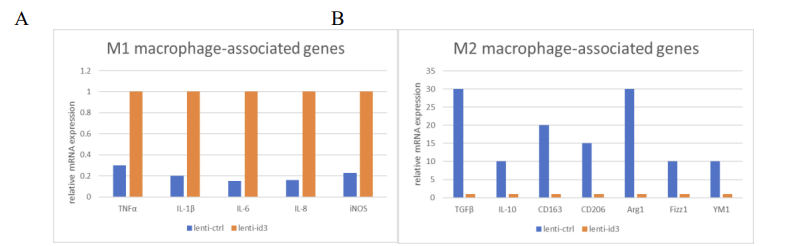
9. Potential challenges and limitations
One potential challenge in this study arises from the specificity and efficiency of the molecular techniques we propose to employe. Although CUT&RUN offers higher resolution and less background noise than ChIP, it still relies heavily on antibody quality. If antibodies against E2A or ELK1 exhibit cross-reactivity or poor binding efficiency, the enrichment data for TBK1 promoter binding might be ambiguous or misleading.
Additionally, E-box or ELK1-binding motifs predicted by deep learning models require experimental validation, as computational predictions may overestimate functional binding. Similarly, lentivirus-based shRNA knockdown systems are prone to off-target gene suppression and variable knockdown efficiency, especially in primary cells like BMDMs or KCs. Residual expression of E2A or ELK1 in knockdown models might still be sufficient to exert regulatory effects, thus complicating interpretation.
Moreover, overexpression of ID3 in immune cells could activate stress responses or off-target transcriptional programs, leading to macrophage dysfunction or cell death. This is especially critical in vivo, where immune balance and systemic responses might amplify subtle cellular perturbations. The in vivo model itself also presents limitations. Injection of modified BMDMs may not fully replicate endogenous macrophage recruitment, differentiation, and function. The tumor microenvironment is highly complex, and changes in phagocytosis or polarization may be influenced by multiple cytokines or stromal interactions beyond the ID3-TBK1 axis.
Furthermore, sex-based differences in immune response could influence macrophage behavior and tumor progression, so both male and female mice should be included and analyzed separately.
10. Alternative experimental approaches
To further support the proposed mechanism, multiple complementary strategies are recommended. For transcriptional regulation, ChIP-seq can provide genome-wide evidence of ID3-E2A/ELK1 binding patterns, while the direct binding ability and transcriptional repression function of the mutant TBK1 promoter to transcription factors validated through electrophoretic mobility shift analysis (EMSA) and luciferase reporter gene detection. Instead of shRNA, CRISPRi or inducible CRISPR-Cas9 knockout systems offer more precise and tunable gene regulation with fewer off-target effects. Moreover, integrating single-cell RNA sequencing technology with CITE seq (a cell indexing technique that simultaneously detects transcriptome and surface proteins) could yield a high-resolution map of macrophage states, enabling simultaneous profiling of transcriptional and surface protein markers.
Spatial transcriptomics or multiplex immunofluorescence may also help visualize the localization and polarization status of macrophages in situ within the tumor. Bioinformatics tools such as motif enrichment analysis, ATAC-seq for chromatin accessibility, and databases like ENCODE and JASPAR[22] can be used to cross-validate predicted regulatory elements. Together, these techniques will enhance the mechanistic rigor and biological relevance of the study.
11. Discussion
Here, we propose a series of experiments aimed at elucidating the mechanistic role of the ID3-E2A/ELK1 complex in regulating TBK1 expression and macrophage polarization within the tumor microenvironment (TME), with implications for antitumor immunity. We anticipate that the complex represses TBK1 transcription through direct binding to its promoter; TBK1 inhibition drives macrophage polarization toward the M1 phenotype, while ID3 enhances phagocytic activity via TBK1 downregulation, thereby reinforcing antitumor immune responses. Notable exceptions include ELK1’s context-dependent functionality—acting as a transcriptional activator when paired with alternative cofactors—and tumor-type specificity, where distinct TME features may modulate the axis’s efficacy. Additionally, compensatory pathways (e.g., STAT6) could counteract TBK1 inhibition, constraining M1 polarization. These exceptions arise from the inherent transcriptional plasticity of immune cells and the redundancy of immune regulatory networks.
Our proposal expands upon the findings of Deng et al., who demonstrated that the ID3 complex enhances macrophage phagocytosis by repressing SIRPA, by positing TBK1 as another key regulatory target that extends the functional scope of the ID3-E2A/ELK1 module. In doing so, results from our proposal might not only confirm ID3 as a critical factor in the anti-tumor activity of macrophages but also provides an upstream transcriptional mechanism for the phenomenon reported by Li et al., where TBK1 deficiency shifts macrophages toward an M1 phenotype through preferential IRF5 activation. Our anticipate results might integrate well with Zhang et al.’s demonstration of TRIM21-mediated post-translational control of M2 drivers, suggesting that macrophage polarization is governed by layered transcriptional and post-translational pathways.
Theoretically, this research establishes a novel ID3-TBK1-IRF5 regulatory axis, reframing TBK1 from a canonical innate signaling kinase into a transcriptionally repressible checkpoint and clarifying the unique, cell-type-dependent anti-tumor function of ID3 within macrophages, even in the face of reports linking high ID3 to poor prognosis in other contexts. Practically, these insights could guide the development of ID3-mimetic drugs or engineered macrophage therapies that leverage ID3 overexpression, provide a mechanistic basis for the use of TBK1 inhibitors as TAM-reprogramming agents, and establish the expression level of ID3 in TAM as a potential biomarker for predicting patients' response to immunotherapy.
The ID3-E2A/ELK1 complex inhibits TBK1 expression by binding to the promoter region of TBK1. This conclusion is supported by evidence such as the prediction of E-Box/ELK1 binding sites through deep learning analysis, the verification of binding through CUT&RUN experiments, and the shRNA knockdown of E2A/ELK1 leading to increased TBK1 expression. ID3-mediated inhibition of TBK1 enhances the phagocytic ability of macrophages, as evidenced by higher tumor cell phagocytosis rates in macrophages overexpressing ID3 in vitro, and the inhibition of tumor growth by injecting ID3-overexpressing BMDM in the Clec4fcreId3f/fmouse model. ID3 promotes M1 macrophage polarization by inhibiting TBK1, as demonstrated by the upregulation of M1 markers (TNFα, IL-1β, IL-6, IL-8, iNOS) and the downregulation of M2 markers (TGFβ, IL-10, CD163, CD206, Arg1, Fizz1, YM1) in ID3-overexpressing cells. These results suggest that targeting the ID3-TBK1 axis may become a new strategy for cancer immunotherapy by promoting M1 polarization and enhancing phagocytosis.
In summary, by identifying TBK1 as a novel regulatory target, our proposed experiments are designed to expand on the functional scope of the ID3-E2A/ELK1 module and clarifies the cell-specific anti-tumor role of ID3 in macrophages. Additionally, we highlight three putative clinical translation directions for cancer immunotherapy. Despite limitations such as the context-dependent function of ELK1, our proposal still offers a new target axis for advancing cancer immunotherapy, laying a foundation for addressing the issue of tumor microenvironment (TME) immunosuppression and improving therapeutic efficacy.
Acknowledgment
All the authors contributed equally to this work and should be considered as co-first author.
References
[1]. Kiri, S., & Ryba, T. (2024). Cancer, metastasis, and the epigenome. Molecular cancer, 23(1), 154.
[2]. Bray, F., Laversanne, M., Sung, H., Ferlay, J., Siegel, R. L., Soerjomataram, I., & Jemal, A. (2024). Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians, 74(3), 229–263.
[3]. Zhang, Y., & Zhang, Z. (2020). The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cellular & molecular immunology, 17(8), 807–821.
[4]. Pittet MJ et al. Clinical relevance of tumour-associated macrophages. Nat Rev Clin Oncol. 2022; 19: 402–21.
[5]. Mantovani A, et al. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017; 14: 399–416.
[6]. Yang Q et al. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm Sin B. 2020; 10: 2156–70.
[7]. Kumari N et al. Tumor-associated macrophages in cancer: recent advancements in cancer nanoimmunotherapies. J Exp Clin Cancer Res. 2022; 41: 68.
[8]. Wei Li et al.Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and EMT in gastric cancer Cell Death & Disease 918 (2019)
[9]. Yubei He et al. The Effects of Hypoxia on the Immune–Metabolic Interplay in Liver Cancer Biomolecules. 2024 Aug 17; 14(8): 1024.
[10]. Sun, Y., Revach, O. Y., Anderson, S., Kessler, E. A., Wolfe, C. H., Jenney, A., Mills, C. E., Robitschek, E. J., Davis, T. G. R., Kim, S., Fu, A., Ma, X., Gwee, J., Tiwari, P., Du, P. P., Sindurakar, P., Tian, J., Mehta, A., Schneider, A. M., Yizhak, K., … Jenkins, R. W. (2023). Targeting TBK1 to overcome resistance to cancer immunotherapy. Nature, 615(7950), 158–167.
[11]. Yuan, J., Pan, J., Zhang, X. et al. TRIM21 reduces H1N1-induced inflammation and apoptosis by regulating the TBK1–IRF3 signaling pathway in A549 cells. Arch Virol 169, 74 (2024).
[12]. Alomari M. (2021). TRIM21 - A potential novel therapeutic target in cancer. Pharmacological research, 165, 105443.
[13]. Gago da Graça, C., Sheikh, A. A., Newman, D. M., Wen, L., Li, S., Shen, J., Zhang, Y., Gabriel, S. S., Chisanga, D., Seow, J., Poch, A., Rausch, L., Nguyen, M. T., Singh, J., Su, C. H., Cluse, L. A., Tsui, C., Burn, T. N., Park, S. L., Von Scheidt, B., … Utzschneider, D. T. (2025). Stem-like memory and precursors of exhausted T cells share a common progenitor defined by ID3 expression. Science immunology, 10(103), eadn1945.
[14]. Chen, Y. S., Aubee, J., DiVito, K. A., Zhou, H., Zhang, W., Chou, F. P., Simbulan-Rosenthal, C. M., & Rosenthal, D. S. (2015). Id3 induces an Elk-1-caspase-8-dependent apoptotic pathway in squamous carcinoma cells. Cancer medicine, 4(6), 914–924.
[15]. Li, Y., Ji, L., Liu, C., Li, J., Wen, D., Li, Z., Yu, L., Guo, M., Zhang, S., Duan, W., Yi, L., Bi, Y., Bu, H., Li, C., & Liu, Y. (2024). TBK1 is involved in M-CSF-induced macrophage polarization through mediating the IRF5/IRF4 axis. The FEBS journal, 291(23), 5214–5235.
[16]. Deng, Z., Loyher, P. L., Lazarov, T., Li, L., Shen, Z., Bhinder, B., Yang, H., Zhong, Y., Alberdi, A., Massague, J., Sun, J. C., Benezra, R., Glass, C. K., Elemento, O., Iacobuzio-Donahue, C. A., & Geissmann, F. (2024). The nuclear factor ID3 endows macrophages with a potent anti-tumour activity. Nature, 626(8000), 864–873.
[17]. Zhang, R., Shen, Y., Zhang, Q., Feng, X., Liu, X., Huo, X., Sun, J., & Hao, J. (2023). TRIM21-mediated Sohlh2 ubiquitination suppresses M2 macrophage polarization and progression of triple-negative breast cancer. Cell death & disease, 14(12), 850.
[18]. Guo Z, Li H, Han M, Xu T, Wu X, Zhuang Y. Modeling Sjögren's syndrome with Id3 conditional knockout mice. Immunol Lett. 2011; 135(1-2): 34-42.
[19]. Sakai M, Troutman TD, Seidman JS, et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity. 2019; 51(4): 655-670.e8.
[20]. Zhong Y, Macgregor-Das A, Saunders T, et al. Mutant p53 Together with TGFβ Signaling Influence Organ-Specific Hematogenous Colonization Patterns of Pancreatic Cancer. Clin Cancer Res. 2017; 23(6): 1607-1620.
[21]. Fornes O, Castro-Mondragon JA, Khan A, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020; 48(D1): D87-D92.
[22]. Liu GH, Tan XY, Xu ZY, Li JX, Zhong GH, Zhai JW, Li MY. REEP3 is a potential diagnostic and prognostic biomarker correlated with immune infiltration in pancreatic cancer. Sci Rep. 2024 Jun 15; 14(1): 13834.
Cite this article
Han,H.;Ruan,C.;Yao,N.;Ye,J.;Zhang,Y. (2025). ID3-E2A/ELK1 Complex Inhibits TBK1 Expression to Promote M1 Macrophage Polarization in the Tumor Microenvironment. Theoretical and Natural Science,144,1-12.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of ICBioMed 2025 Symposium: AI for Healthcare: Advanced Medical Data Analytics and Smart Rehabilitation
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Kiri, S., & Ryba, T. (2024). Cancer, metastasis, and the epigenome. Molecular cancer, 23(1), 154.
[2]. Bray, F., Laversanne, M., Sung, H., Ferlay, J., Siegel, R. L., Soerjomataram, I., & Jemal, A. (2024). Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians, 74(3), 229–263.
[3]. Zhang, Y., & Zhang, Z. (2020). The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cellular & molecular immunology, 17(8), 807–821.
[4]. Pittet MJ et al. Clinical relevance of tumour-associated macrophages. Nat Rev Clin Oncol. 2022; 19: 402–21.
[5]. Mantovani A, et al. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017; 14: 399–416.
[6]. Yang Q et al. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm Sin B. 2020; 10: 2156–70.
[7]. Kumari N et al. Tumor-associated macrophages in cancer: recent advancements in cancer nanoimmunotherapies. J Exp Clin Cancer Res. 2022; 41: 68.
[8]. Wei Li et al.Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and EMT in gastric cancer Cell Death & Disease 918 (2019)
[9]. Yubei He et al. The Effects of Hypoxia on the Immune–Metabolic Interplay in Liver Cancer Biomolecules. 2024 Aug 17; 14(8): 1024.
[10]. Sun, Y., Revach, O. Y., Anderson, S., Kessler, E. A., Wolfe, C. H., Jenney, A., Mills, C. E., Robitschek, E. J., Davis, T. G. R., Kim, S., Fu, A., Ma, X., Gwee, J., Tiwari, P., Du, P. P., Sindurakar, P., Tian, J., Mehta, A., Schneider, A. M., Yizhak, K., … Jenkins, R. W. (2023). Targeting TBK1 to overcome resistance to cancer immunotherapy. Nature, 615(7950), 158–167.
[11]. Yuan, J., Pan, J., Zhang, X. et al. TRIM21 reduces H1N1-induced inflammation and apoptosis by regulating the TBK1–IRF3 signaling pathway in A549 cells. Arch Virol 169, 74 (2024).
[12]. Alomari M. (2021). TRIM21 - A potential novel therapeutic target in cancer. Pharmacological research, 165, 105443.
[13]. Gago da Graça, C., Sheikh, A. A., Newman, D. M., Wen, L., Li, S., Shen, J., Zhang, Y., Gabriel, S. S., Chisanga, D., Seow, J., Poch, A., Rausch, L., Nguyen, M. T., Singh, J., Su, C. H., Cluse, L. A., Tsui, C., Burn, T. N., Park, S. L., Von Scheidt, B., … Utzschneider, D. T. (2025). Stem-like memory and precursors of exhausted T cells share a common progenitor defined by ID3 expression. Science immunology, 10(103), eadn1945.
[14]. Chen, Y. S., Aubee, J., DiVito, K. A., Zhou, H., Zhang, W., Chou, F. P., Simbulan-Rosenthal, C. M., & Rosenthal, D. S. (2015). Id3 induces an Elk-1-caspase-8-dependent apoptotic pathway in squamous carcinoma cells. Cancer medicine, 4(6), 914–924.
[15]. Li, Y., Ji, L., Liu, C., Li, J., Wen, D., Li, Z., Yu, L., Guo, M., Zhang, S., Duan, W., Yi, L., Bi, Y., Bu, H., Li, C., & Liu, Y. (2024). TBK1 is involved in M-CSF-induced macrophage polarization through mediating the IRF5/IRF4 axis. The FEBS journal, 291(23), 5214–5235.
[16]. Deng, Z., Loyher, P. L., Lazarov, T., Li, L., Shen, Z., Bhinder, B., Yang, H., Zhong, Y., Alberdi, A., Massague, J., Sun, J. C., Benezra, R., Glass, C. K., Elemento, O., Iacobuzio-Donahue, C. A., & Geissmann, F. (2024). The nuclear factor ID3 endows macrophages with a potent anti-tumour activity. Nature, 626(8000), 864–873.
[17]. Zhang, R., Shen, Y., Zhang, Q., Feng, X., Liu, X., Huo, X., Sun, J., & Hao, J. (2023). TRIM21-mediated Sohlh2 ubiquitination suppresses M2 macrophage polarization and progression of triple-negative breast cancer. Cell death & disease, 14(12), 850.
[18]. Guo Z, Li H, Han M, Xu T, Wu X, Zhuang Y. Modeling Sjögren's syndrome with Id3 conditional knockout mice. Immunol Lett. 2011; 135(1-2): 34-42.
[19]. Sakai M, Troutman TD, Seidman JS, et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity. 2019; 51(4): 655-670.e8.
[20]. Zhong Y, Macgregor-Das A, Saunders T, et al. Mutant p53 Together with TGFβ Signaling Influence Organ-Specific Hematogenous Colonization Patterns of Pancreatic Cancer. Clin Cancer Res. 2017; 23(6): 1607-1620.
[21]. Fornes O, Castro-Mondragon JA, Khan A, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020; 48(D1): D87-D92.
[22]. Liu GH, Tan XY, Xu ZY, Li JX, Zhong GH, Zhai JW, Li MY. REEP3 is a potential diagnostic and prognostic biomarker correlated with immune infiltration in pancreatic cancer. Sci Rep. 2024 Jun 15; 14(1): 13834.