







1. Introduction
Epilepsy is a kind of chronic brain disease and the pathogenesis is complicated. Epidemiological investigation shows that there are about 60million epileptic patients in the world at present. Epilepsy has a high incidence rate and disability rate, causing heavy medical and health care burden to society and families. As the specific pathogenesis of epilepsy is still unclear, epilepsy cannot be completely cured. In addition, about 75% of epilepsy patients in the world cannot receive timely and effective treatment. The current clinical treatments are mainly controlling seizures but there is no medicine to cure the disease. What’s more, some people gain resistance toward certain medicine. Therefore, it is one of the urgent problems to keep on studying the pathogenesis of epilepsy and at the same time, seek an ideal treatment.
2. Sodium channel
Epilepsy's onset and progression are intimately correlated with the structural and functional alterations of ion channels, and the formation and transmission of the action potential depend heavily on the voltage-gated sodium channel (VGSC or Nav) [1]. Many current studies show that numerous syndromes of epilepsy are relevant to the gene of Na v.
Voltage-gated sodium channel is a kind of transmembrane protein that widely exist in the excitable cells of whole body, consisting of one big alpha subunit and two beta small subunits. Alpha subunit, which is considered as functional subunit, is made up of about 2000 amino acids and is highly conservative. It was found that there was a single expression of the alpha subunit which has the channel activity on oocytes of a kind of African toad, but the beta subunit does not have that activity without alpha subunit [2]. Therefore, alpha subunit is mainly focused on in the research of Nav.
There are nine different isomers of alpha subunit (from Nav1.1 to Nav1.9) in mammal animal which are coded by different genes. The change in the structures of those genes are highly relevant to the occurrence of epilepsy [3]. The first found sodium channel gene related to epilepsy was SCN1B. The mutated gene leads to the change of beta subunit by destroying the disulfide bond of a domain of it. In 1998, Wallace reported a genetic mutation finding about beta subunit in a family of generalized epilepsy with febrile seizures plus (GEFS+). In 2009, the homozygous mutation of SCN1B was found in the check on sixty-seven patients of Dravet disease. The mutation influences mutual interaction between beta subunit and adhesion molecule, which caused over-excitation of neurons.
Among all the gene coding Nav, SCN1A has the largest number of mutations related to epilepsy, and SCN1A is also considered to be the one most closely related to epilepsy [4]. About 85% of Dravet Syndrome patients have mutations in SCN1A gene, and the most common phenotype of SCN1A gene mutations (about 70%) is also DS [5]. Moreover, there are other genes from SCN family related to epilepsy. The mutation of SCN2A was found in the patients with benign familial neonatal seizures and the mutation of SCN5A was found in an epilepsy patient with QT syndrome but the concrete mechanism is not clear. The researches on the relationship between SCN3A mutation and epilepsy started later than others. Until 2008, Holland found the mutation is in the connected area of the four mentioned domains and the mutation decreases the threshold of the action potential. Interestingly, different from other genes, the mutation of SCN8A will increase the threshold of action potential thereby reducing the sensitivity to epilepsy according to the experiment done by Martin [6].
3. Calcium CIUM ion channel
Calcium ion channel, a kind of glycoprotein or lateral glycosylated protein, is widely located in different tissues of the human body. It can be divided into ligand-gated and voltage-gated. The pathway of calcium ion influx mainly depends on voltage-gated calcium channels (VGCCs) which can be subdivided into high voltage activated (HVA) and low voltage-activated (LVA). According to physiological characteristics, VGCCs can be divided into different types like N,T and so on. T-type calcium channels play a crucial role in the reticular concussion circuit composed of thalamic relay neurons, cortical pyramidal cells, and reticular colliculus nuclei [7]. As shown in Fig 1.
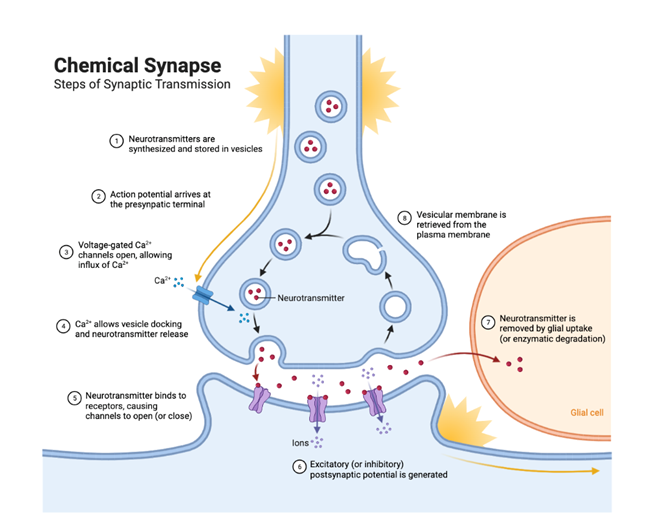
Figure 1. The function of calcium ion in synaptic transmission [7].
T-type calcium channels, belonging to LVA, consist of alpha-one subunit. Important electrophysiological characteristics of T-type calcium channels: (1) joining the form of spike-and-wave discharges, SWDs. The low amplitude depolarization induced by N-Methyl-D-aspartate receptor can produce a waterfall effect, opening a large number of T-type calcium channels and forming low threshold calcium spikes. T-type calcium channels participate in the formation of low threshold calcium spikes, triggering SWDs, leading to the occurrence of epilepsy [8]. (2) being involved in thalamus-cortex neuron synchronization concussion which can occur in sleep under physiological conditions. Abnormal super synchronization discharge is closely related to childhood absence epilepsy (CAE). (3) inducing window- current. The inactivation and activation voltage ranges of T-type calcium channels almost coincide, and some channels that are activated at the membrane potential level produce window currents, which can influence the excitability of neurons.
According to the difference of alpha subunit, T-type calcium channels are divided into three series: Ca v1, Cav2 and Cav3, and Cav3. Cav3 series includes Cav3.1, Cav3.2, and Cav3.3. The experiment done by Ernst W L [9] shows that the density of calcium ion current in the relayed nucleus of thalamus increases when Cav3.1 over-expresses thereby inducing spike waves which can cause epilepsy. CACNA1H, coding Cav3.2, when mutating, can increase calcium ion influx, which will cause the accumulation of calcium ions. Therefore, the neurons will be over-excited and epilepsy will happen. The mutation body of CACNA1H can be found in Temporal Lobe Epilepsy, Childhood Absence Epilepsy and so on. Currently, the relationship between epilepsy and Cav3.3 is not clear.
4. HCN channel
HCN, the abbreviation of hyperpolarization-activated cyclic nucleotide-gated, is a crucial channel in the occurrence of epilepsy which widely exists in the thalamus and hippocampus. It is encoded by four genes (HCN1~4) and except for HCN3, the rest three genes are directly related to epilepsy. HCN structurally forms tetrameric subunits, each of which has a topology of six transmembrane domains (s1~s6)(Fig 2). HCN channel can produce a special current named hyperpolarization activation current (Ih) and the nature of Ih differs from location and number of HCN channel. It is a special kind of voltage-gated ion channel, which is one of the few voltage-gated channels that open at resting potential so that Ih current plays a basic role in controlling resting membrane potential and neuron-specific discharge activity [10]. Ih current can regulate excitatory and inhibitory postsynaptic potentials of cortical and hippocampal neurons and also control the tonic discharge of neurons in cerebellar and pacesetter activity of neurons in subcortical areas [11], which is the basis of focal seizures and generalized seizures. When neurons are depolarized by synaptic input, the depolarized sodium current mediated by HCN channel will be closed, and the channel will be inactivated by depolarization, which makes the membrane potential hyperpolarize back to the resting state; On the contrary, hyperpolarization input will activate IH current and repolarize neurons back to a resting state.
The mutation of HCN channel is closely related to epilepsy and in the genetic creening of epilepsy patients, numerous mutations of HCN channel and its subunit can be found. Bonzanni found a mutation of HCN1 in generalized epilepsy patients and this mutation increases the discharge rate of neurons as well as excitability [12]. The mutation of HCN2 will lead to deactivation of the channel and then the excitability of neurons will increase obviously. According to the research done by Rivolta [13], Two infant boys who had benign myoclonic seizures were revealed to have an HCN4 mutation, which increases neuronal excitability and increases the risk of developing epilepsy. All the mutation usually causes channel function damage, affecting normal function and eventually leading to epilepsy. HCN channel also co-operates with helper protein that regulates its normal functions and the change of helper protein can lead to the change of Ih current, which will lead to the occurrence of epilepsy. TRIP8b is one of the most studied helper proteins. In the trip8b knockout mouse model, a large number of HCN channels in dendritic cells were lost, and the model shows epileptic syndrome.
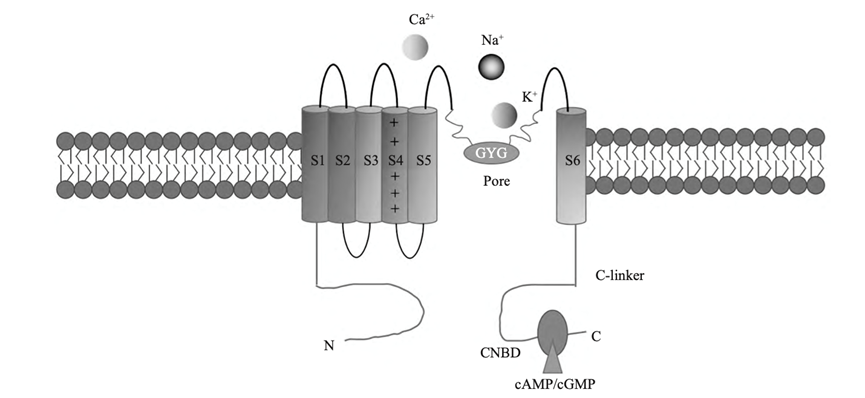
Figure 2. Structure of HCN channel [14].
5. Potassium ion channel
Potassium channels are the largest and most structurally diverse in the ion channel family, encoded by more than 80 human genes. Potassium channel regulates cell function extensively by selectively controlling the flow of K+ inside and outside cells and among all types of it, two-pore domain K+ are studied most in recent years.
In the genetic group of humans, there are 15 genes related to K2P, and according to the structure and function, K2P can be divided into 6 sub-groups. Among them, TREK and TASK families are mainly concentrated on.
There are three numbers in TREK family: TREK-1, TREK-2, TRAAK(KCNK4). Although there is nearly 80% homology among the three, the specific paralysor of TREK-1 and its analogues do not affect TREK-2 and TRAAK. TREK-1 is highly expressed in human CNS, especially in the presynaptic and postsynaptic parts of cortex,hippocampus, and thalamus, which are sensitive brain areas for epilepsy [15]. TREK-1 is regulated by many factors, such as mechanical changes, temperature, pH changes and other physical stimuli, polyunsaturated fatty acids and phospholipids and other chemicals [16]. According to the research, TREK1-removed mice are more likely to show the status of epilepsy which is produced by KA and PTZ. The current reports related to TREK-2 are very limited so the concrete relationship between it and epilepsy is limited. Based on the experiments, there is a conjecture [17]: The post-transcriptional mechanism involved in non-coding RNA plays an important role in TREK-2 regulation, and TREK-2 up-regulated after epileptic injury may play a protective role in the maintenance of potassium and glutamate levels in astrocytes. Different from other K2P channels, KCN4 is almost only expressed in central and peripheral neurons and the retina, so the multidirectional effects caused by mutation are unexpected. The function of KCNK4 channel mutant increased significantly, which lead that the K+ channel continuing to open, the basic K+ current increasing significantly, and the depolarization current increasing significantly. Therefore, a series of serious results will appear.
TASK1~3 are expressed in normal hippocampal pyramidal neurons and dentate gyrus granulosa cells. At the subcellular level, TASK-1 is expressed around the dendrites and nuclei of human hippocampal pyramidal neurons and dentate gyrus granulosa cells; TASK-3 is located in the nucleus of pyramidal cells and granulosa cells rather than dendrites. In the classical gerbil model of hereditary epilepsy, TASK-1 is not only expressed in hippocampal neurons, but also astrocytes. The up-regulation of TASK-1 in astrocytes of epilepsy-prone gerbils may lead to the increase of extracellular K+ level by increasing K+ outward rectification, which increases cell excitability. After seizures, the rapid down-regulation of TASK-1 in astrocytes is part of the rapid adaptation process. TASK-2 is expressed in all layers of hippocampus and dentate gyrus, and it is a non-inactivated channel. Kim reported that temporal lobe epilepsy in rats is related to the increased expression of TASK-2 in hippocampus and dentate gyrus [18]. TASK-3 channel is relevant to numerous disorders, including sleep/wakefulness control, and epilepsy. TASK-3 reduction can reduce the action potential threshold, resulting in prolonged excitability of hippocampal neurons in patients with temporal lobe epilepsy.
6. KCI co-transporter 2 (KCC2) and epilepsy
The potassium chloride cotransporters (KCCS) is a branch of the cation chloride cotransporter family. So far, four subtypes of KCCS(KCC1-KCC4) have been identified, and above them, KCC2 is the only subunit that only expresses in neurons. KCC2 regulates the outward flow of Cl- in the neuronal plasma membrane and makes a great contribution in neurotransmission by reducing the concentration of Cl- in cells γ- Aminobutyric acid (GABA) and glycine.
It has been proved that KCC2 mRNA is highly expressed in the neurons of the whole CNS. KCC2 is the main Cl- outflow promoter in the mature brain which exerts rapid hyperpolarization inhibition by reducing intracellular cl- concentration and activating GABAA receptor [19]. During development, GABAergic response experienced a transition from excitability to inhibition. This transition is determined by the concentration of Cl- in cells and the developmental regulation of Cl- in cells is related to the incremental regulation of KCC2, which is that the expression level of KCC2 was low at birth and peaked in the first few weeks after birth. When KCC2 is not enough or lost, Cl- homeostasis in neurons disintegrates, the excitability of neurons is excessively enhanced, and the susceptibility to seizures is increased.
The decline of KCC2 expression has been found in a variety of in vivo and in vitro animal models and human intractable epilepsy studies. The decrease of KCC2 changes the homeostasis of Cl- in neurons and changes the GABAergic response in the direction of depolarization, so that the excitability of neurons is excessively increased and the function of neurons is damaged, leading to the occurrence of epilepsy. In the patch clamp technique, it was found that GABA and glycine have an inhibitory function in the neurons of wild-type mice. On the contrary, in KCC2-removed mice, the two neurotransmitters have an excitatory function. Complete knockout of KCC2 gene leads to abnormal muscle tone due to the abnormal excitatory activity of GABA and glycine after birth, which will lead to death. Moreover, studies on mice that knocked out exon 1 of KCC2 have found that mice are easy to induce frequent systemic seizures by mild stimulation [20].
7. GABA and glutamate
The imbalance between excitation and inhibition of the central nervous system is the main mechanism of epilepsy. Among them, the main representative of excitatory neurotransmitters in the brain is glutamate, while the main representative of inhibitory neurotransmitters is γ- Aminobutyric acid( γ- aminobutyric acid,GABA). Under physiological conditions, extracellular glutamate in central nervous system is mainly regulated by the Glu-Gln cycle. Once the regulation is abnormal, a large amount of extracellular glutamate will be accumulated, then epilepsy may occur.
Glu-Gln cycle is an important way to maintain the normal metabolism of glutamate in the central nervous system (Fig 3). Glutamate released to synaptic space is mainly absorbed by glutamate transporter on the membrane of astrocyte and some of the absorbed glutamates takes part in the tricarboxylic acid cycle while the rest will be turned into glutamine.
The glutamate transporter is critical for the Glu-Gln cycle as well as for the control of glutamate and GABA. Excitatory amino acid transporters (EAATs) in human body are divided into five subtypes: EAAT1-5 and among them GLT-1(EAAT2) and GLAST (EAAT1) are mainly expressed in astrocyte. GLT-1 is a sodium ion-dependent transmembrane transporter, widely distributed in CNS. GLT-1 is in charge of the ingestion of over 90% glutamate in the brain and abnormal GLT-1 will not only destroy the level of glutamate, making a large amount of glutamate accumulate in the synaptic space but also indirectly affect the synthesis of GABA. Selective knockout of the gene encoding GLT-1 in mouse astrocytes revealed a significant reduction in glutamate uptake, increased mortality, weight loss, and seizures of epilepsy [21]. Detection of brain tissue in patients who have temporal lobe epilepsy showed that the level of GLT-1 protein in hippocampus and temporal lobe decreased significantly [22]. All the above experiments show that GLT-1 is closely related to epilepsy. GLAST is mainly expressed in cerebellum and cerebral cortex. Similar to GLT-1, GLAST is also a sodium-dependent transmembrane symporter. However, there is something different from GLT-1. GLAST also plays a significant part in preventing neuronal excitatory injury. Studies have shown that although lack of GLAST will not directly lead to spontaneous seizures, lack of GLAST will increase the duration and severity of seizures and shorten the incubation period of seizures. There is few researchers since EAAT4 and EAAT5 do not have a significant impact on how epilepsy is regulated.
Extracellular glutamate is mainly cleared by GLT-1 and GLAST expressed on astrocytes. In contrast, the neuronal transporter EAAT3 does not seem to have a strong scavenging effect on extracellular glutamate, but the expression and functional changes of EAAT3 are also closely related to epilepsy. EAAT3 regulates glutamate concentration by providing glutamate for GABA synthesis to inhibitory neurons and limiting the movement of glutamate to adjacent synapses.
Astrocytes are the largest glial cells and the most abundant cell type in the brain. When pathological changes occur in central nervous system, astrocytes will be activated, and corresponding changes will occur in gene expression, cell morphology, cell function, and other levels [23]. At the same time, the number of cells will also change. This process is called reactive astrocyte proliferation. This proliferation may have beneficial or harmful effects on peripheral nerve cells. Moreover, some researches show that after the damage to central nervous system, the function of astrocyte will be reduced or lost due to this proliferation, which will lead to a declined ingestion of glutamate and then excitotoxicity may occur. Whereas there are also studies showing that reactive astrocyte proliferation can enhance extracellular glutamate metabolism, release neurotrophic factors and prevent oxidative stress, thus protecting neurons.
8. Gaba receptor
GABA receptors can be divided into three types: GABAA, GABAB and GABAC. The first two kinds of receptors are widely expressed in the human CNS while GABAC can be found in retina. Therefore, when studying diseases like epilepsy, GABAA and GABAB are mainly focused on.
Most GABAA receptors consist of at least one α and β and γ form. GABAA receptor consisting of Alpha1, Beta two, and Gama (GABRA1) is the most in the brain, especially in hippocampus and cerebral cortex. GABAA, an ionotropic receptor, after being activated, can promote chloride outflow. Some studies [24] have shown that in temporal lobe tissue of brain of epilepsy patients, the expression of GABRA1 declined but in frontal lobe of brain, the expression is not distinguishable from normal people, which illustrates that the change of GABAA receptor expression in different tissues of patients with epilepsy is not a single change. GABAB, a metabotropic receptor, after activated, GABAB2 subunit and GABAB1 will form a complex. They exert biological effects through cAMP signaling pathway in nerve cells, inhibiting Ca2 + channels, reducing the permeability of Ca2+ and opening the potassium channel on presynaptic and postsynaptic membrane thereby producing inhibitory postsynaptic potential further inhibiting the release of neurotransmitters from the presynaptic membrane. Molecular biological experiments found that the relative expression of GABAB protein in hippocampus of rats in each group showed that the relative expression of GABAB protein in epileptic rats was significantly lower than that in normal rats [25]. So, when gene coding the receptor protein mutates, the inhibitory functions are weakened or lost, and the inhibition of neurons is greatly reduced, which will cause occurrence of epilepsy.
9. Others
Human infection with parasitic diseases can affect the central nervous system and then lead to epilepsy. People especially in rural villages easy to get an infection due to the custom. Neurocysticercosis (NCC) is one of the most common infective diseases. Human, as the definitive host of Taenia solium Linnaeus, is easy to get infected when eating raw pork. The larva in the raw pork grow up in the human body and release cysticercus cellulose to different parts of body. Epilepsy caused by cysticercus infection is related to the direct invasion of brain tissue by cysticercus. Soon after the larva invades the brain parenchyma, it gradually enters the degenerative process and transforms into mineralized nodules due to the immune attack of the host. With the calcification of Cysticercus, the proliferation of its peripheral glia and the residual antigenic material remain in the brain parenchyma, changing to a persistent epileptogenic lesion. In addition to Cysticercosis, CNS-infected parasitic diseases can also be seen in toxoplasmosis. Tachyzoites of toxoplasmosis can quickly proliferate and spread to all the organs including the brain. Epilepsy caused by toxoplasma infection is not only related to the cyst formed in brain tissue but also related to the positive serum antibody. According to research [26], the positive rate of IgG in patients with CNS disorder was 19.81%, higher than in normal people (only 5.42%). Therefore, it shows that the mechanism of epilepsy caused by toxoplasma is related to an immune reaction.
Schistosomiasis is also a common parasitic disease. Nervous schistosomiasis is one of the most serious manifestations of schistosomiasis. Patients with neurological schistosomiasis usually get epilepsy, headache, and papilledema. Similar to toxoplasma, epilepsy caused by Schistosoma is not only related to the occupation of the egg or larva but also the immune reaction of the host. Some studies focusing on the gut flora also show that the microbiota-gut-brain axis is related to epilepsy in some ways.
10. Conclusion
Epilepsy is a disease with complex etiology and is difficult to cure, which seriously affects the quality of life of patients. The sodium, potassium, calcium, and HCN ion channels on the membrane are crucial for preserving the excitability of neurons. When genes coding those channels mutate, epilepsy occurs due to the change of excitability. The imbalance between excitatory and inhibitory functions is also related to epilepsy. If the synthetic amount of GABA declines due to the lack of raw material (glutamate) or the receptor of GABA changes, the inhibitory function will reduce and epilepsy may happen. Moreover, there are also other risk factors like parasite infection and imbalance of gut flora related to epilepsy. To sum up, epilepsy is a kind of chronic disease with complex pathogeny and serious consequences. The review of pathogeny of epilepsy is helpful in so many fields like prevention, clinical medicine and so on. To have a clearer understanding of epilepsy, more experiments are required.
References
[1]. D Jiang, J Zhang, Z Xia. Structural Advances in Voltage-Gated Sodium Channels. Front Pharmacol. 2022 Jun 3;13:908867.
[2]. Y Zhou, Z Luo, B Xiao. Voltage-gated Sodium Channels and Epilepsy [J]. Journal of Apoplexy and Nervous Diseases,2015, 32(12):1142-1143.
[3]. K.L.Helbig , E.M.Goldberg . SCN3A-Related Neurodevelopmental Disorder. 2021 Jun 3 [updated 2021 Nov 4]. In: M.P.Adam, G.M.Mirzaa , R.A.Pagon, et.al®[Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022.
[4]. E Amadori, G Pellino , L Bansal , et al. Genetic paroxysmal neurological disorders featuring episodic ataxia and epilepsy. Eur J Med Genet. 2022 Apr; 65(4):104450.
[5]. R Guerrini. Dravet syndrome: the main issues[J]. European Journal of Paediatric Neurology: EJPN: Of icial Journal of the European Paedia- tric Neurology Society,2012, 1( 16) : S1-4.
[6]. I. A.Ademuwagun , S.O.Rotimi , S Syrbe , et,al. Voltage Gated Sodium Channel Genes in Epilepsy: Mutations, Functional Studies, and Treatment Dimensions. Front Neurol. 2021 Mar 24;12:600050.
[7]. G.W. Zamponi, P Lory, E Perez-Reyes.Role of voltage- gated calcium channels in epilepsy[J].Pflugers Arch, 2010, 460(2): 395-403.
[8]. W.L.Ernst , Y Zhang, J.W.Yoo, et al.Genetic enhancement of thalamocortical network activity by elevating 1G-mediated low voltage activated calcium current induces pure absence epilepsy[J]. J Neurosci, 2009, 29(6): 1615-1625.
[9]. X Li, B Qin, M Huang ,et al . in the Pathogenesis of Epilepsy involved HCN Channel [J]. Chemistry of life,2021, 41(09): 1961-1966.
[10]. J.C.DiFrancesco, B Castellotti, R Milanesi , et al. HCN ion channels and accessory proteins in epilepsy: genetic analysis of a large cohort of patients and review of the literature. Epilepsy Res. 2019 Jul; 153:49-58.
[11]. M Bonzanni , J.C.Difrancesco , R Milanesi , et al. A novel de novo HCN1 loss-of-function mutation in genetic generalized epilepsy causing increased neuronal excitability. Neurobiol Dis, 2018, 118: 55-63
[12]. E Eren-Koçak ,T Dalkara . Ion Channel Dysfunction and Neuroinflammation in Migraine and Depression. Front Pharmacol. 2021 Nov 10;12:777607.
[13]. Q Yang , P Kuzyk , I Antonov , et al. Hyperpolarization- activated, cyclic nucleotide-gated cation channels in Aplysia: contribution to classical conditioning [J]. Proc Natl Acad Sci USA, 2015, 112(52): 16030-5.
[14]. A Djillani , J Mazella , C Heurteaux ,et al . Role of TREK-1 in Health and Disease, Focus on the Central Nervous System. Front Pharmacol. 2019 Apr 11; 10:379.
[15]. M Lengyel , P Enyedi , G Czirják . Negative Influence by the Force: Mechanically Induced Hyperpolarization via K2P Background Potassium Channels. Int J Mol Sci. 2021 Aug 23;22(16):9062.
[16]. A Mathie , E.L.Veale , A Golluscio , R.G.Holden , Y Walsh . Pharmacological Approaches to Studying Potassium Channels. Handb Exp Pharmacol. 2021; Exp Pharmacol. 2021; 267:83-111.
[17]. C Steriade , J French ,et.al . Epilepsy: key experimental therapeutics in early clinical development. Expert Opin Investig Drugs. 2020 Apr; 29(4):373-383.
[18]. M.A.Virtanen , P Uvarov , M Mavrovic , et.al . The Multifaceted Roles of KCC2 in Cortical Development. Trends Neurosci. 2021 May;44(5):378-392.
[19]. G Di Cristo, P.N.Awad , S Hamidi , et.al . KCC2, epileptiform synchronization, and epileptic disorders. Prog Neurobiol. 2018 Mar; 162:1-16.
[20]. G.L.Sarlo , K.F.Holton . Brain concentrations of glutamate and GABA in human epilepsy: A review. Seizure. 2021 Oct; 91:213-227.
[21]. R.S.Duman , G Sanacora , J.H.Krystal . Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron. 2019 Apr 3; 102(1):75-90.
[22]. G.A.Czapski , J.B.Strosznajder . Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer's Disease. Int J Mol Sci. 2021 Oct 28; 22(21):11677.
[23]. A Sood , K Preeti , V Fernandes , et.al. Glia: A major player in glutamate-GABA dysregulation- mediated neurodegeneration. J Neurosci Res. 2021 Dec; 99(12):3148-3189.
[24]. R.W.Olsen . GABAA receptor: Positive and negative allosteric modulators. Neuropharmacology. 2018 Jul 1;136(Pt A):10-22.
[25]. C Schulte, H.M. Maric. Expanding GABAAR pharmacology via receptor-associated proteins. Curr Opin Pharmacol. 2021 Apr; 57:98-106.
[26]. T.S. Lima , M.B.Lodoen . Mechanisms of Human Innate Immune Evasion by Toxoplasma gondii. Front Cell Infect Microbiol. 2019 Apr16; 9:103
Cite this article
Sun,K. (2023). The Pathogenesis of Epilepsy. Theoretical and Natural Science,3,368-376.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Biological Engineering and Medical Science (ICBioMed 2022), Part I
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. D Jiang, J Zhang, Z Xia. Structural Advances in Voltage-Gated Sodium Channels. Front Pharmacol. 2022 Jun 3;13:908867.
[2]. Y Zhou, Z Luo, B Xiao. Voltage-gated Sodium Channels and Epilepsy [J]. Journal of Apoplexy and Nervous Diseases,2015, 32(12):1142-1143.
[3]. K.L.Helbig , E.M.Goldberg . SCN3A-Related Neurodevelopmental Disorder. 2021 Jun 3 [updated 2021 Nov 4]. In: M.P.Adam, G.M.Mirzaa , R.A.Pagon, et.al®[Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022.
[4]. E Amadori, G Pellino , L Bansal , et al. Genetic paroxysmal neurological disorders featuring episodic ataxia and epilepsy. Eur J Med Genet. 2022 Apr; 65(4):104450.
[5]. R Guerrini. Dravet syndrome: the main issues[J]. European Journal of Paediatric Neurology: EJPN: Of icial Journal of the European Paedia- tric Neurology Society,2012, 1( 16) : S1-4.
[6]. I. A.Ademuwagun , S.O.Rotimi , S Syrbe , et,al. Voltage Gated Sodium Channel Genes in Epilepsy: Mutations, Functional Studies, and Treatment Dimensions. Front Neurol. 2021 Mar 24;12:600050.
[7]. G.W. Zamponi, P Lory, E Perez-Reyes.Role of voltage- gated calcium channels in epilepsy[J].Pflugers Arch, 2010, 460(2): 395-403.
[8]. W.L.Ernst , Y Zhang, J.W.Yoo, et al.Genetic enhancement of thalamocortical network activity by elevating 1G-mediated low voltage activated calcium current induces pure absence epilepsy[J]. J Neurosci, 2009, 29(6): 1615-1625.
[9]. X Li, B Qin, M Huang ,et al . in the Pathogenesis of Epilepsy involved HCN Channel [J]. Chemistry of life,2021, 41(09): 1961-1966.
[10]. J.C.DiFrancesco, B Castellotti, R Milanesi , et al. HCN ion channels and accessory proteins in epilepsy: genetic analysis of a large cohort of patients and review of the literature. Epilepsy Res. 2019 Jul; 153:49-58.
[11]. M Bonzanni , J.C.Difrancesco , R Milanesi , et al. A novel de novo HCN1 loss-of-function mutation in genetic generalized epilepsy causing increased neuronal excitability. Neurobiol Dis, 2018, 118: 55-63
[12]. E Eren-Koçak ,T Dalkara . Ion Channel Dysfunction and Neuroinflammation in Migraine and Depression. Front Pharmacol. 2021 Nov 10;12:777607.
[13]. Q Yang , P Kuzyk , I Antonov , et al. Hyperpolarization- activated, cyclic nucleotide-gated cation channels in Aplysia: contribution to classical conditioning [J]. Proc Natl Acad Sci USA, 2015, 112(52): 16030-5.
[14]. A Djillani , J Mazella , C Heurteaux ,et al . Role of TREK-1 in Health and Disease, Focus on the Central Nervous System. Front Pharmacol. 2019 Apr 11; 10:379.
[15]. M Lengyel , P Enyedi , G Czirják . Negative Influence by the Force: Mechanically Induced Hyperpolarization via K2P Background Potassium Channels. Int J Mol Sci. 2021 Aug 23;22(16):9062.
[16]. A Mathie , E.L.Veale , A Golluscio , R.G.Holden , Y Walsh . Pharmacological Approaches to Studying Potassium Channels. Handb Exp Pharmacol. 2021; Exp Pharmacol. 2021; 267:83-111.
[17]. C Steriade , J French ,et.al . Epilepsy: key experimental therapeutics in early clinical development. Expert Opin Investig Drugs. 2020 Apr; 29(4):373-383.
[18]. M.A.Virtanen , P Uvarov , M Mavrovic , et.al . The Multifaceted Roles of KCC2 in Cortical Development. Trends Neurosci. 2021 May;44(5):378-392.
[19]. G Di Cristo, P.N.Awad , S Hamidi , et.al . KCC2, epileptiform synchronization, and epileptic disorders. Prog Neurobiol. 2018 Mar; 162:1-16.
[20]. G.L.Sarlo , K.F.Holton . Brain concentrations of glutamate and GABA in human epilepsy: A review. Seizure. 2021 Oct; 91:213-227.
[21]. R.S.Duman , G Sanacora , J.H.Krystal . Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron. 2019 Apr 3; 102(1):75-90.
[22]. G.A.Czapski , J.B.Strosznajder . Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer's Disease. Int J Mol Sci. 2021 Oct 28; 22(21):11677.
[23]. A Sood , K Preeti , V Fernandes , et.al. Glia: A major player in glutamate-GABA dysregulation- mediated neurodegeneration. J Neurosci Res. 2021 Dec; 99(12):3148-3189.
[24]. R.W.Olsen . GABAA receptor: Positive and negative allosteric modulators. Neuropharmacology. 2018 Jul 1;136(Pt A):10-22.
[25]. C Schulte, H.M. Maric. Expanding GABAAR pharmacology via receptor-associated proteins. Curr Opin Pharmacol. 2021 Apr; 57:98-106.
[26]. T.S. Lima , M.B.Lodoen . Mechanisms of Human Innate Immune Evasion by Toxoplasma gondii. Front Cell Infect Microbiol. 2019 Apr16; 9:103