







1. Introduction
The CRISPR–Cas system represents the core adaptive immune mechanism in bacteria and archaea. It functions by capturing and integrating foreign DNA fragments (spacers) into the CRISPR array, which are then transcribed into CRISPR RNAs (crRNAs) and assembled with Cas proteins to form interference complexes. These complexes recognize and cleave complementary invading nucleic acids, thereby providing precise protection against phages and plasmids [1-3]. Since its first demonstration for genome editing in eukaryotes in 2012, the CRISPR–Cas system has revolutionized gene therapy, molecular diagnostics, and crop improvement [4-9].
Structurally and functionally, the CRISPR–Cas system comprises two main components: the CRISPR array, which acts as a “memory” repository of invader sequences [10]; and Cas effector proteins, which serve as the “execution” machinery for target cleavage [11]. Based on effector composition, CRISPR–Cas systems are classified into Class 1 (multi-protein effectors) and Class 2 (single-protein effectors). Class 2 effectors such as Cas9 (Type II), Cas12 (Type V), and Cas13 (Type VI) have been widely adopted for DNA or RNA targeting applications due to their simplicity and programmability [12-19].
Despite their importance, only a limited number of Cas proteins have been reliably annotated, and the diversity of naturally occurring Cas variants in microbial genomes far exceeds current annotations. Many novel Cas proteins exhibit substantial sequence divergence from known families, leading to poor sensitivity of traditional sequence-alignment tools (e.g., BLAST, HMMER) against these divergent variants [20,21]. For example, multiple novel CasX/CasY systems identified in environmental metagenomes were often missed by standard alignment methods due to low sequence similarity [22]; similarly, newly discovered Class 2 systems from uncultivated microbes showed markedly reduced detection rates with HMMER-based searches [23]. Moreover, evolutionary analyses have highlighted that the rapid emergence of new Cas subtypes outpaces template-based alignment approaches [24]. Consequently, there is a pressing need for intelligent algorithms that jointly leverage sequence and structural information for accurate Cas protein identification.
Recent advances in protein language models (e.g., ESM, ProtBERT, TAPE) have demonstrated powerful capability in capturing sequence semantics and inferring structural and functional properties. Notably, ESM-2 performs large-scale unsupervised sequence modeling [25]; ESMFold uses ESM-2 latent representations to predict protein structures directly [26]; and ESM-3 further integrates structural information into the training process to achieve joint sequence–structure representation learning [27]. These developments open new avenues for multi-modal protein analysis.
In this work, we introduce ESMFusion, a multi-modal deep learning framework for high-accuracy Cas protein identification. ESMFusion integrates sequence embeddings derived from ESM-2 with structural embeddings obtained from ESMFold and ESM-3, combining them into a unified representation. A Multi-Scale Convolutional Neural Network (MSCNN) is employed to capture both local and global feature patterns, followed by a multilayer perceptron (MLP) classifier for final prediction. Comprehensive comparative experiments demonstrate that ESMFusion substantially outperforms traditional and single-modality baselines, achieving a classification accuracy of 93.37% on curated datasets.
2. Materials and methods
2.1. Overall framework of ESMFusion
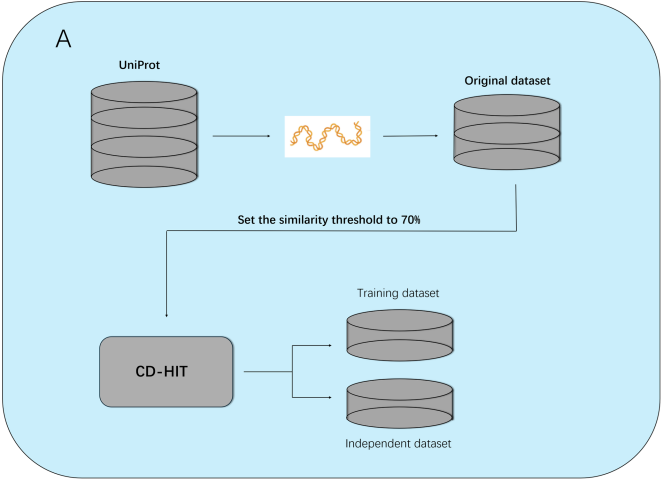
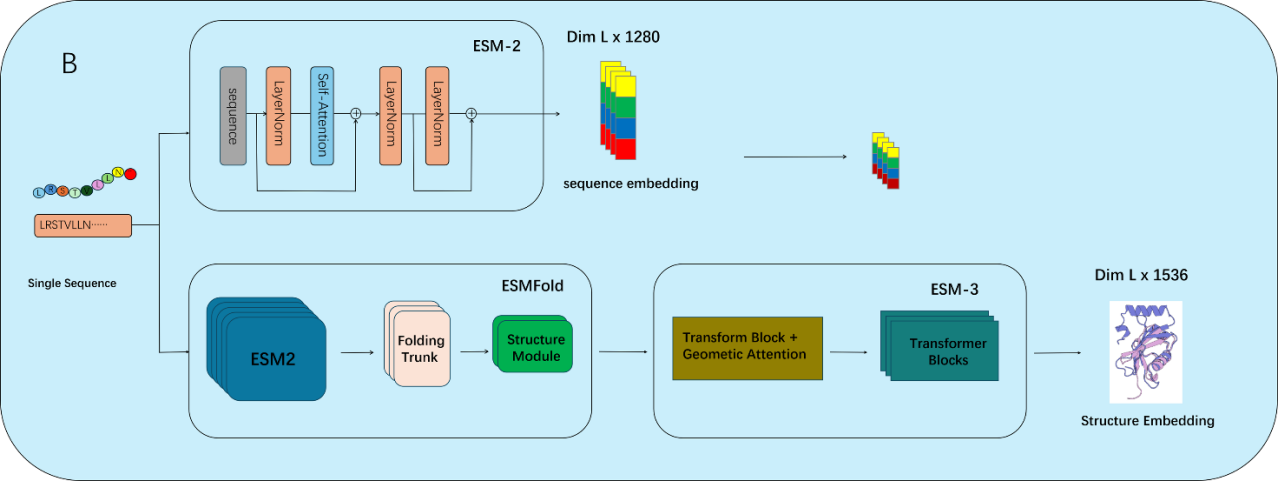
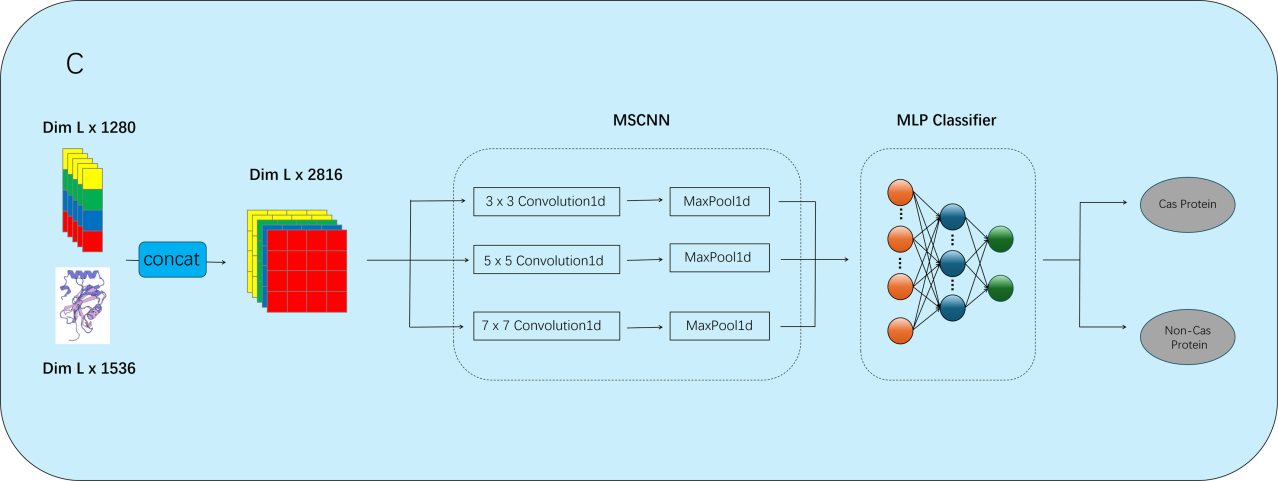
As illustrated in Figures 1–3, ESMFusion comprises three main components: dataset construction, feature representation, and classification. First (Figure 1), we curated a comprehensive benchmark dataset comprising diverse Cas and non-Cas protein sequences to support robust model training and evaluation. Special attention was paid to ensuring representative sequence diversity while minimizing redundancy. Next (Figure 2), protein representations were extracted from both sequence and structural modalities. For sequence encoding, we utilized the pretrained ESM-2 (650M) model and applied mean pooling over the final hidden layer to obtain a 1280-dimensional feature vector. For structural encoding, the protein’s 3D structure was first predicted using ESMFold, and embeddings were then generated using ESM-3, yielding a 1536-dimensional vector. These two representations were concatenated to form a unified 2816-dimensional feature vector. Finally (Figure 3), the concatenated embedding was fed into a multi-scale convolutional neural network (MSCNN) composed of multiple parallel one-dimensional convolutional branches with varying kernel sizes to capture multi-scale semantic patterns. The outputs were pooled, concatenated, and flattened into a single vector, which was passed through a two-layer fully connected neural network. A sigmoid activation function was applied to produce the final binary prediction, indicating whether the input corresponds to a Cas or non-Cas protein.
2.2. Dataset introduction
To establish a robust and dependable predictive framework, we constructed a curated benchmark dataset for model training and performance evaluation. Initially, Cas protein sequences were retrieved from the UniProt [28] database using the keyword “CRISPR-associated,” and only manually reviewed entries were retained as the preliminary positive set. To mitigate sequence redundancy and avoid overfitting, we applied CD-HIT [29] with a 70% sequence identity cutoff, retaining a single representative protein from each cluster. This process yielded a final positive dataset consisting of 209 non-redundant Cas protein sequences.
For the negative class, we randomly selected 209 manually reviewed non-Cas protein sequences based on a previously published study [30]. Sequences containing non-standard amino acid residues (such as ‘X’, ‘B’, or ‘Z’) were excluded, and CD-HIT was again applied to eliminate sequences with more than 40% internal similarity. Furthermore, to ensure that no negative sample shared more than 70% sequence identity with any positive sample, the entire dataset underwent an additional round of clustering at the 70% threshold. This procedure ensured that both positive and negative sets were free from excessive homology.
The final dataset was split into two subsets: a training set (Cas300) and an independent test set (Cas118). Cas300 consisted of 150 Cas and 150 non-Cas proteins, while Cas118 included 59 proteins for each class, facilitating reliable model evaluation on unseen data.
2.3. Traditional single descriptors
To systematically characterize protein sequences, We employed nine representative single-feature descriptors from the iLearnPlus toolkit [31], categorized into three types: composition-based, physicochemical property-based, and sequence-order-based features.
2.3.1. Composition-based features
Amino Acid Composition (AAC) [32] captures the frequency of each amino acid, providing a global view of sequence content. CKSAAP [33] encodes the frequency of amino acid pairs separated by k residues (k = 3 in this study), reflecting local residue co-occurrence.
2.3.2. Physicochemical property-based features
PAAC incorporates physicochemical properties and sequence order into AAC. APAAC extends PAAC by including amphiphilic attributes such as hydrophobicity and polarity, enhancing functional representation.
2.3.3. Sequence-order-based features
Normalized Moreau–Broto Autocorrelation (NMBroto) calculates autocorrelation values based on physicochemical indices, highlighting long-range residue interactions. CTDC and CTDT [34] group amino acids by specific properties and compute their composition and transition frequencies across the sequence. Quasi-Sequence Order (QSOrder) [35] integrates sequence-order effects with property-based distances between residues. Adaptive Skip Dipeptide Composition (ASDC) [36] extends dipeptide encoding by including non-adjacent residue pairs, improving representation of distant correlations relevant to structural context.
3. Result
3.1. Performance comparison of traditional single descriptors
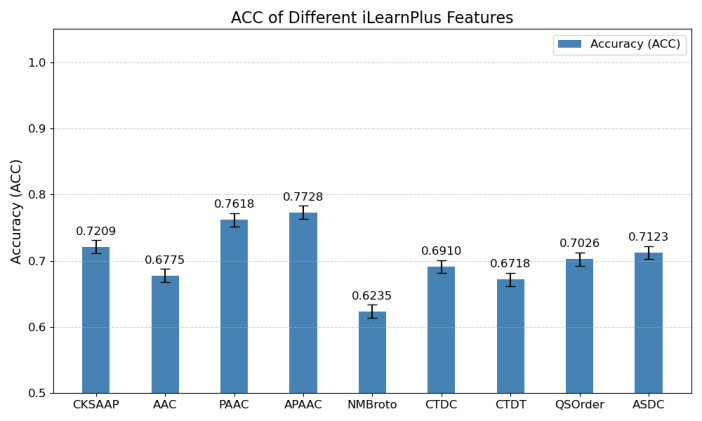
To assess the classification performance of traditional handcrafted features in Cas protein identification, we evaluated nine representative single-feature descriptors introduced in Section 2.3 on the Cas300. Each descriptor was used to encode the sequences and fed into an identical classification model to ensure a fair comparison. Classification accuracy was employed as the primary evaluation metric.
As illustrated in Figure 4, descriptors based on physicochemical properties—PAAC and APAAC—achieved relatively high performance, both exceeding 75% accuracy. In contrast, composition-based descriptors (AAC and CKSAAP) produced moderate results, with accuracies around 70%. Sequence-order-based descriptors, such as CTDC, CTDT, and NMBroto, generally underperformed, with accuracies falling below 70% in most cases.
3.2. Performance comparison of ESM-2 models with different scales
In this experiment, we evaluated the performance of four different scales of ESM-2 (8M, 35M, 150M, and 650M) in generating sequence embeddings and performing Cas protein classification tasks.
To better understand the impact of model size on classification performance, we first applied UMAP (Uniform Manifold Approximation and Projection) to reduce the dimensionality of the positive and negative sequence embeddings generated by each model. As shown in Figure 5, the UMAP results reveal a clear trend: with the increase in model size, the distinction between positive and negative samples becomes more apparent. This suggests that larger ESM-2 models are capable of capturing more complex and subtle patterns in protein sequences, contributing to more robust classification performance.
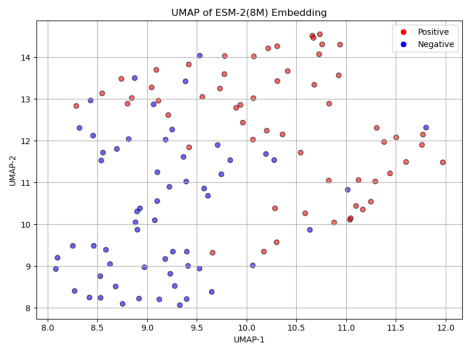
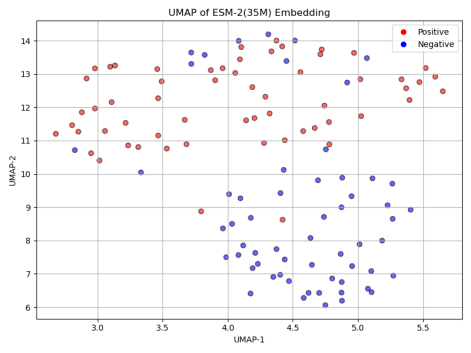
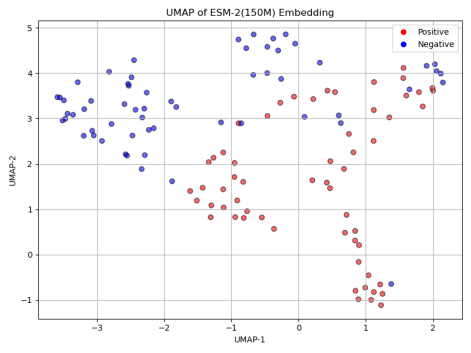
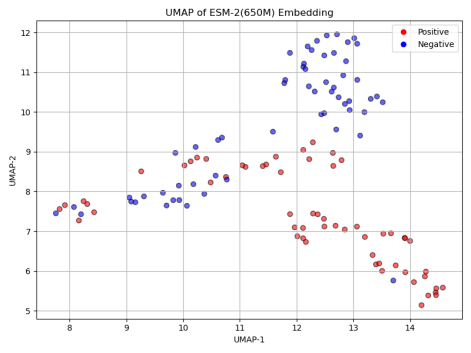
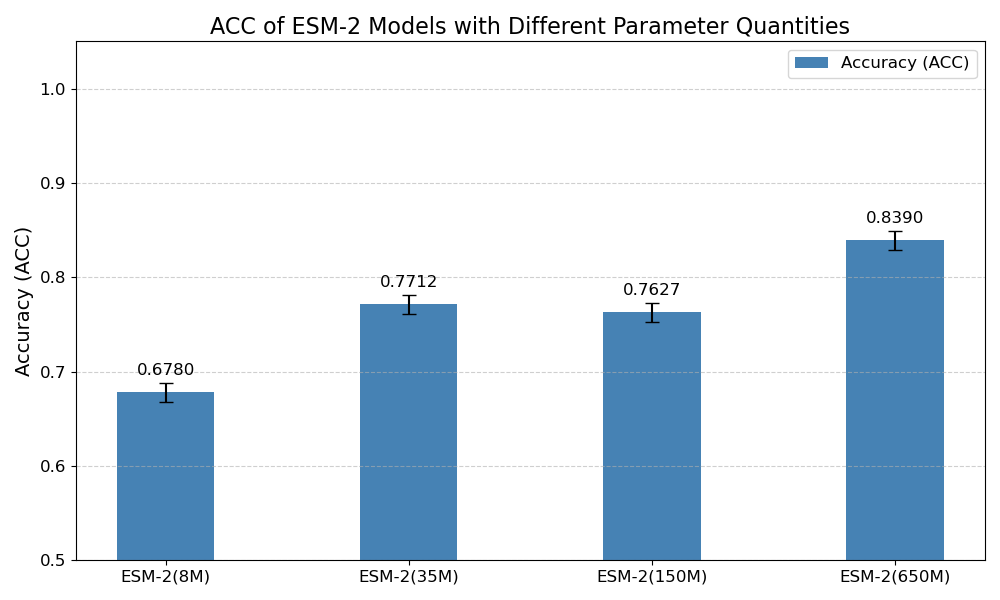
Next, we assessed the classification accuracy of each model on the Cas300 dataset, which contains both Cas and non-Cas protein samples. As shown in the results in Figure 6, the classification accuracy significantly improved with the increase in model parameters. Specifically, ESM-2 (8M) model, the smallest in terms of parameters, achieved the lowest accuracy at only 67.80%. This indicates that the model's capacity is limited, preventing it from capturing the complex features within the sequence data. ESM-2 (35M) and ESM-2 (150M) models showed similar performance, with accuracy rates of 77.12% and 76.27%, respectively. Although these models performed better than ESM-2 (8M), their accuracies still did not exceed 80%, suggesting that medium-scale models have certain limitations in classification performance.
In contrast, ESM-2 (650M) model outperformed the others, achieving an accuracy of 83.90%, significantly higher than the other three models. This result indicates that larger ESM-2 models are better at capturing subtle relationships within protein sequences, leading to a significant improvement in classification performance. Given the outstanding performance of ESM-2 (650M) model in the classification task, we decided to use this model for subsequent experiments to further validate the advantages of large-scale pre-trained models in Cas protein classification.
3.3. Comparison of different models for cas protein classification performance
In this experiment, we evaluated the performance of various models in the Cas protein classification task, including the original ESM-2, fine-tuned ESM-2, ESM-3, CASPredict, CRISPRCasStack, and our proposed ESMFusion model. The objective of this experiment was to assess the classification accuracy of different model architectures and analyze the advantages and limitations of each model in the Cas protein classification task.
The classification accuracies of the models are shown in Figure 7. The results are as follows: the original ESM-2 model achieved an accuracy of 83.90%, demonstrating relatively good performance. The ESM-3 model slightly outperformed ESM-2, with an accuracy of 84.75%. The fine-tuned ESM-2 model achieved an accuracy of 72.08%, indicating that while fine-tuning can optimize the model for a specific task, its performance was not as good as the original pre-trained ESM-2 model. The CASPredict [30], specifically designed for CRISPR-Cas classification, achieved an accuracy of 84.84%, slightly higher than ESM-3. As the current state-of-the-art model for Cas protein prediction, CRISPRCasStack [37] achieved the highest accuracy of 94.07%, outperforming all other models. ESMFusion, which combines the outputs of ESM-2 and ESM-3, achieved an accuracy of 89.14%. Although this result shows a significant improvement over single models, it still falls short of CRISPRCasStack.
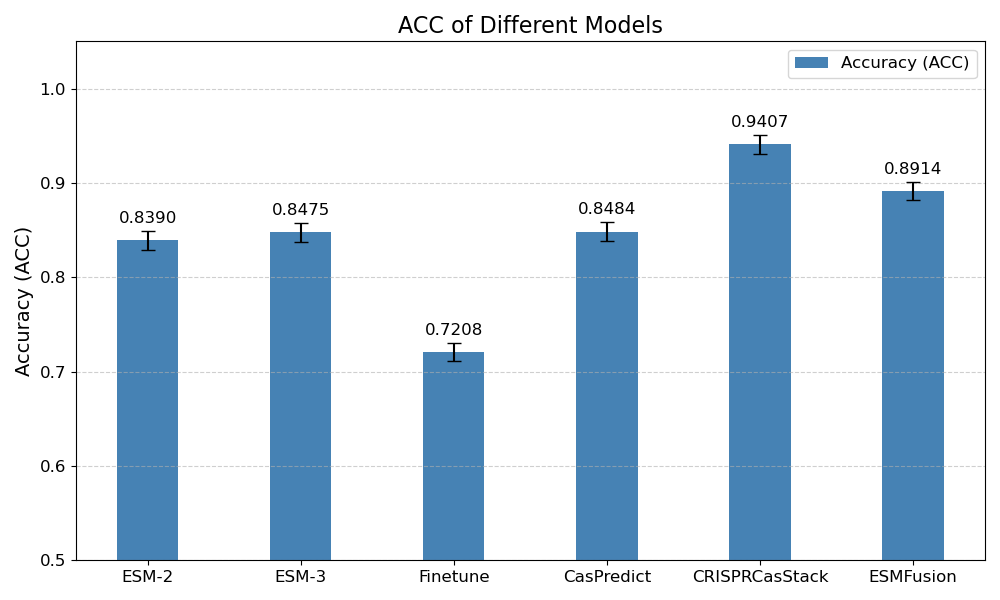
The results of this experiment demonstrate that combining multiple models and integrating sequence and structural information can significantly enhance classification accuracy. While CRISPRCasStack exhibited the highest accuracy, ESMFusion model also showed strong classification performance, achieving an accuracy of 89.14%, which is superior to the single ESM-2 and ESM-3. However, it is still slightly lower than CRISPRCasStack, indicating that task-specific optimized models continue to have a certain advantage in performance.
3.4. Improvement of ESMFusion with MSCNN for cas protein classification
In this experiment, we further enhanced the performance of ESMFusion model by incorporating MSCNN to better capture multi-scale feature representations. The addition of MSCNN allows the model to extract hierarchical and multi-resolution patterns from the protein sequence embeddings, which has been shown to improve the classification accuracy.
As described in the previous experiments, the baseline ESMFusion model, which combines the outputs from both ESM-2 and ESM-3 models, achieved a classification accuracy of 89.14%. However, by introducing MSCNN, which utilizes multiple convolutional kernels of different sizes to capture both local and global features, we observed a significant improvement in the model's performance.
The MSCNN architecture consists of three convolutional layers with kernel sizes of 3, 5, and 7, followed by max-pooling operations to reduce dimensionality. The multi-scale features are then concatenated and passed through a MLP for final classification. This architecture is designed to extract multi-scale information from protein sequence embeddings and provide more nuanced feature representations for classification.
After integrating MSCNN into ESMFusion, we re-evaluated its classification accuracy on the Cas300. As shown in Figure 8, the accuracy increased to 93.37%, which is comparable to the performance of the CRISPRCasStack (94.07%), the current state-of-the-art method for Cas protein classification. This improvement demonstrates the effectiveness of multi-scale feature extraction in enhancing classification accuracy, making the MSCNN-augmented ESMFusion model a strong contender for Cas protein classification tasks.
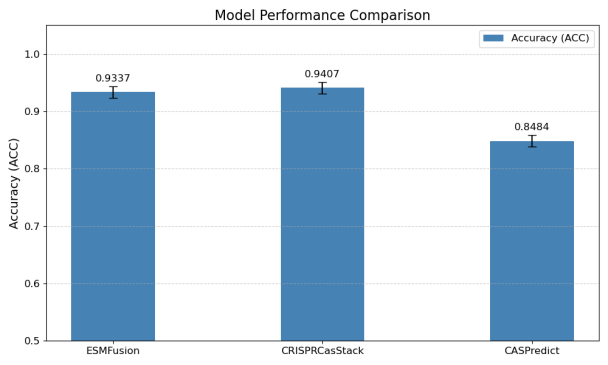
These findings highlight the importance of incorporating multi-scale convolutional techniques into large pre-trained models for protein sequence classification. The MSCNN's ability to capture fine-grained patterns across different scales helps the model generalize better to complex biological data, ultimately achieving near-state-of-the-art performance.
4. Disscussion
In this study, we proposed ESMFusion, a multimodal deep learning framework that integrates protein sequence and structural information for high-precision Cas protein classification. By combining embeddings from ESM-2, ESMFold, and ESM-3, and enhancing them via a Multi-Scale CNN, ESMFusion effectively captures both global and local biological features.
We first evaluated ESM-2 of varying scales, with the 650M model showing the best performance (83.90%) and serving as our baseline. Comparative experiments demonstrated that ESMFusion (89.14%) outperformed single-modality and existing methods. Adding MSCNN further boosted accuracy to 93.37%, matching state-of-the-art tools like CRISPRCasStack.
In summary, ESMFusion shows strong generalization ability and classification performance on the Cas protein prediction task. The experimental results highlight the effectiveness of integrating multimodal biological features, and the proposed approach provides new insights and methodologies for future protein function prediction tasks based on pretrained models. In future work, we plan to incorporate additional structural predictors, explore graph neural networks, and conduct cross-species generalization studies to further improve the robustness and biological interpretability of the model.
References
[1]. B. J. Rauch et al., “Inhibition of CRISPR-Cas9 with Bacteriophage Proteins, ” Cell, vol. 168, no. 1–2, pp. 150-158.e10, Jan. 2017, doi: 10.1016/j.cell.2016.12.009.
[2]. R. Sorek, V. Kunin, and P. Hugenholtz, “CRISPR — a widespread system that provides acquired resistance against phages in bacteria and archaea, ” Nat. Rev. Microbiol., vol. 6, no. 3, pp. 181–186, Mar. 2008, doi: 10.1038/nrmicro1793.
[3]. G. Vestergaard, R. A. Garrett, and S. A. Shah, “CRISPR adaptive immune systems of Archaea, ” RNA Biol., vol. 11, no. 2, pp. 156–167, Feb. 2014, doi: 10.4161/rna.27990.
[4]. B. M. Hussen et al., “Targeting miRNA by CRISPR/Cas in cancer: advantages and challenges, ” Mil. Med. Res., vol. 10, no. 1, p. 32, 2023.
[5]. J. Lou et al., “The CRISPR-Cas system as a tool for diagnosing and treating infectious diseases, ” Mol. Biol. Rep., vol. 49, no. 12, pp. 11301–11311, 2022.
[6]. N. Rangel, V. Camargo, G. Castellanos, M. Forero-Castro, and M. Rondón-Lagos, “Exploring the advantages and limitations of CRISPR-cas in breast cancer, ” Gene Expr., vol. 23, no. 2, pp. 116–126, 2024.
[7]. E. B. Rukavtsova, N. S. Zakharchenko, V. G. Lebedev, and K. A. Shestibratov, “CRISPR-Cas genome editing for horticultural crops improvement: advantages and prospects, ” Horticulturae, vol. 9, no. 1, p. 38, 2022.
[8]. C. Li, E. Brant, H. Budak, and B. Zhang, “CRISPR/Cas: a Nobel Prize award-winning precise genome editing technology for gene therapy and crop improvement, ” J. Zhejiang Univ.-Sci. B, vol. 22, no. 4, pp. 253–284, 2021.
[9]. A. Din, M. A. Wani, C. Jin, I. T. Nazki, J. Ma, and F. Li, “Post-genomic era of CRISPR/Cas technology in ornamental plants: advantages, limitations, and prospects, ” Ornam. Plant Res., vol. 5, no. 1, 2025.
[10]. R. Barrangou et al., “CRISPR provides acquired resistance against viruses in prokaryotes, ” Science, vol. 315, no. 5819, pp. 1709–1712, 2007.
[11]. M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna, and E. Charpentier, “A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity, ” science, vol. 337, no. 6096, pp. 816–821, 2012.
[12]. K. S. Makarova, F. Zhang, and E. V. Koonin, “SnapShot: class 2 CRISPR-Cas systems, ” Cell, vol. 168, no. 1, pp. 328–328, 2017.
[13]. E. Chaudhary, A. Chaudhary, S. Sharma, V. Tiwari, and M. Garg, “Different Classes of CRISPR-Cas Systems, ” in Gene Editing in Plants: CRISPR-Cas and Its Applications, Springer, 2024, pp. 73–94.
[14]. K. S. Makarova et al., “An updated evolutionary classification of CRISPR–Cas systems, ” Nat. Rev. Microbiol., vol. 13, no. 11, pp. 722–736, 2015.
[15]. S. Shmakov et al., “Diversity and evolution of class 2 CRISPR–Cas systems, ” Nat. Rev. Microbiol., vol. 15, no. 3, pp. 169–182, 2017.
[16]. M. Asmamaw and B. Zawdie, “Mechanism and applications of CRISPR/Cas-9-mediated genome editing, ” Biol. Targets Ther., pp. 353–361, 2021.
[17]. E. Charpentier and L. A. Marraffini, “Harnessing CRISPR-Cas9 immunity for genetic engineering, ” Curr. Opin. Microbiol., vol. 19, pp. 114–119, 2014.
[18]. Y. Yang, D. Wang, P. Lü, S. Ma, and K. Chen, “Research progress on nucleic acid detection and genome editing of CRISPR/Cas12 system, ” Mol. Biol. Rep., vol. 50, no. 4, pp. 3723–3738, 2023.
[19]. D. B. Cox et al., “RNA editing with CRISPR-Cas13, ” Science, vol. 358, no. 6366, pp. 1019–1027, 2017.
[20]. S. F. Altschul, W. Gish, W. Miller, E. W. Myers, and D. J. Lipman, “Basic local alignment search tool, ” J. Mol. Biol., vol. 215, no. 3, pp. 403–410, 1990.
[21]. S. R. Eddy, “Accelerated profile HMM searches, ” PLoS Comput. Biol., vol. 7, no. 10, p. e1002195, 2011.
[22]. D. Paez-Espino et al., “Uncovering Earth’s virome, ” Nature, vol. 536, no. 7617, pp. 425–430, 2016.
[23]. D. Burstein et al., “New CRISPR–Cas systems from uncultivated microbes, ” Nature, vol. 542, no. 7640, pp. 237–241, 2017.
[24]. K. S. Makarova et al., “Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants, ” Nat. Rev. Microbiol., vol. 18, no. 2, pp. 67–83, 2020.
[25]. B. Hie et al., “A high-level programming language for generative protein design, ” BioRxiv, pp. 2022–12, 2022.
[26]. Z. Lin et al., “Language models of protein sequences at the scale of evolution enable accurate structure prediction, ” BioRxiv, vol. 2022, p. 500902, 2022.
[27]. T. Hayes et al., “Simulating 500 million years of evolution with a language model, ” Science, p. eads0018, 2025.
[28]. U. Consortium, “UniProt: a hub for protein information, ” Nucleic Acids Res, vol. 43, no. D1, pp. D204–D212, 2015.
[29]. L. Fu, B. Niu, Z. Zhu, S. Wu, and W. Li, “CD-HIT: accelerated for clustering the next-generation sequencing data, ” Bioinformatics, vol. 28, no. 23, pp. 3150–3152, 2012.
[30]. S. Yang, J. Huang, and B. He, “CASPredict: a web service for identifying Cas proteins, ” PeerJ, vol. 9, p. e11887, 2021.
[31]. Z. Chen et al., “iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization, ” Nucleic Acids Res., vol. 49, no. 10, pp. e60–e60, 2021.
[32]. M. Bhasin and G. P. Raghava, “Classification of nuclear receptors based on amino acid composition and dipeptide composition, ” J. Biol. Chem., vol. 279, no. 22, pp. 23262–23266, 2004.
[33]. K. Chen, L. A. Kurgan, and J. Ruan, “Prediction of flexible/rigid regions from protein sequences using k-spaced amino acid pairs, ” BMC Struct. Biol., vol. 7, pp. 1–13, 2007.
[34]. N. Xiao, D.-S. Cao, M.-F. Zhu, and Q.-S. Xu, “protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences, ” Bioinformatics, vol. 31, no. 11, pp. 1857–1859, 2015.
[35]. K.-C. Chou, “Prediction of protein subcellular locations by incorporating quasi-sequence-order effect, ” Biochem. Biophys. Res. Commun., vol. 278, no. 2, pp. 477–483, 2000.
[36]. L. Wei, C. Zhou, H. Chen, J. Song, and R. Su, “ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides, ” Bioinformatics, vol. 34, no. 23, pp. 4007–4016, 2018.
[37]. T. Zhang, Y. Jia, H. Li, D. Xu, J. Zhou, and G. Wang, “CRISPRCasStack: A stacking strategy-based ensemble learning framework for accurate identification of Cas proteins, ” Brief. Bioinform., vol. 23, no. 5, p. bbac335, 2022.
Cite this article
Wang,Z. (2025). ESMFusion: A High-Accuracy Detection Method for Cas Proteins by Integrating Pretrained Language Models. Applied and Computational Engineering,157,92-101.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of CONF-CDS 2025 Symposium: Data Visualization Methods for Evaluatio
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. B. J. Rauch et al., “Inhibition of CRISPR-Cas9 with Bacteriophage Proteins, ” Cell, vol. 168, no. 1–2, pp. 150-158.e10, Jan. 2017, doi: 10.1016/j.cell.2016.12.009.
[2]. R. Sorek, V. Kunin, and P. Hugenholtz, “CRISPR — a widespread system that provides acquired resistance against phages in bacteria and archaea, ” Nat. Rev. Microbiol., vol. 6, no. 3, pp. 181–186, Mar. 2008, doi: 10.1038/nrmicro1793.
[3]. G. Vestergaard, R. A. Garrett, and S. A. Shah, “CRISPR adaptive immune systems of Archaea, ” RNA Biol., vol. 11, no. 2, pp. 156–167, Feb. 2014, doi: 10.4161/rna.27990.
[4]. B. M. Hussen et al., “Targeting miRNA by CRISPR/Cas in cancer: advantages and challenges, ” Mil. Med. Res., vol. 10, no. 1, p. 32, 2023.
[5]. J. Lou et al., “The CRISPR-Cas system as a tool for diagnosing and treating infectious diseases, ” Mol. Biol. Rep., vol. 49, no. 12, pp. 11301–11311, 2022.
[6]. N. Rangel, V. Camargo, G. Castellanos, M. Forero-Castro, and M. Rondón-Lagos, “Exploring the advantages and limitations of CRISPR-cas in breast cancer, ” Gene Expr., vol. 23, no. 2, pp. 116–126, 2024.
[7]. E. B. Rukavtsova, N. S. Zakharchenko, V. G. Lebedev, and K. A. Shestibratov, “CRISPR-Cas genome editing for horticultural crops improvement: advantages and prospects, ” Horticulturae, vol. 9, no. 1, p. 38, 2022.
[8]. C. Li, E. Brant, H. Budak, and B. Zhang, “CRISPR/Cas: a Nobel Prize award-winning precise genome editing technology for gene therapy and crop improvement, ” J. Zhejiang Univ.-Sci. B, vol. 22, no. 4, pp. 253–284, 2021.
[9]. A. Din, M. A. Wani, C. Jin, I. T. Nazki, J. Ma, and F. Li, “Post-genomic era of CRISPR/Cas technology in ornamental plants: advantages, limitations, and prospects, ” Ornam. Plant Res., vol. 5, no. 1, 2025.
[10]. R. Barrangou et al., “CRISPR provides acquired resistance against viruses in prokaryotes, ” Science, vol. 315, no. 5819, pp. 1709–1712, 2007.
[11]. M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna, and E. Charpentier, “A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity, ” science, vol. 337, no. 6096, pp. 816–821, 2012.
[12]. K. S. Makarova, F. Zhang, and E. V. Koonin, “SnapShot: class 2 CRISPR-Cas systems, ” Cell, vol. 168, no. 1, pp. 328–328, 2017.
[13]. E. Chaudhary, A. Chaudhary, S. Sharma, V. Tiwari, and M. Garg, “Different Classes of CRISPR-Cas Systems, ” in Gene Editing in Plants: CRISPR-Cas and Its Applications, Springer, 2024, pp. 73–94.
[14]. K. S. Makarova et al., “An updated evolutionary classification of CRISPR–Cas systems, ” Nat. Rev. Microbiol., vol. 13, no. 11, pp. 722–736, 2015.
[15]. S. Shmakov et al., “Diversity and evolution of class 2 CRISPR–Cas systems, ” Nat. Rev. Microbiol., vol. 15, no. 3, pp. 169–182, 2017.
[16]. M. Asmamaw and B. Zawdie, “Mechanism and applications of CRISPR/Cas-9-mediated genome editing, ” Biol. Targets Ther., pp. 353–361, 2021.
[17]. E. Charpentier and L. A. Marraffini, “Harnessing CRISPR-Cas9 immunity for genetic engineering, ” Curr. Opin. Microbiol., vol. 19, pp. 114–119, 2014.
[18]. Y. Yang, D. Wang, P. Lü, S. Ma, and K. Chen, “Research progress on nucleic acid detection and genome editing of CRISPR/Cas12 system, ” Mol. Biol. Rep., vol. 50, no. 4, pp. 3723–3738, 2023.
[19]. D. B. Cox et al., “RNA editing with CRISPR-Cas13, ” Science, vol. 358, no. 6366, pp. 1019–1027, 2017.
[20]. S. F. Altschul, W. Gish, W. Miller, E. W. Myers, and D. J. Lipman, “Basic local alignment search tool, ” J. Mol. Biol., vol. 215, no. 3, pp. 403–410, 1990.
[21]. S. R. Eddy, “Accelerated profile HMM searches, ” PLoS Comput. Biol., vol. 7, no. 10, p. e1002195, 2011.
[22]. D. Paez-Espino et al., “Uncovering Earth’s virome, ” Nature, vol. 536, no. 7617, pp. 425–430, 2016.
[23]. D. Burstein et al., “New CRISPR–Cas systems from uncultivated microbes, ” Nature, vol. 542, no. 7640, pp. 237–241, 2017.
[24]. K. S. Makarova et al., “Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants, ” Nat. Rev. Microbiol., vol. 18, no. 2, pp. 67–83, 2020.
[25]. B. Hie et al., “A high-level programming language for generative protein design, ” BioRxiv, pp. 2022–12, 2022.
[26]. Z. Lin et al., “Language models of protein sequences at the scale of evolution enable accurate structure prediction, ” BioRxiv, vol. 2022, p. 500902, 2022.
[27]. T. Hayes et al., “Simulating 500 million years of evolution with a language model, ” Science, p. eads0018, 2025.
[28]. U. Consortium, “UniProt: a hub for protein information, ” Nucleic Acids Res, vol. 43, no. D1, pp. D204–D212, 2015.
[29]. L. Fu, B. Niu, Z. Zhu, S. Wu, and W. Li, “CD-HIT: accelerated for clustering the next-generation sequencing data, ” Bioinformatics, vol. 28, no. 23, pp. 3150–3152, 2012.
[30]. S. Yang, J. Huang, and B. He, “CASPredict: a web service for identifying Cas proteins, ” PeerJ, vol. 9, p. e11887, 2021.
[31]. Z. Chen et al., “iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization, ” Nucleic Acids Res., vol. 49, no. 10, pp. e60–e60, 2021.
[32]. M. Bhasin and G. P. Raghava, “Classification of nuclear receptors based on amino acid composition and dipeptide composition, ” J. Biol. Chem., vol. 279, no. 22, pp. 23262–23266, 2004.
[33]. K. Chen, L. A. Kurgan, and J. Ruan, “Prediction of flexible/rigid regions from protein sequences using k-spaced amino acid pairs, ” BMC Struct. Biol., vol. 7, pp. 1–13, 2007.
[34]. N. Xiao, D.-S. Cao, M.-F. Zhu, and Q.-S. Xu, “protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences, ” Bioinformatics, vol. 31, no. 11, pp. 1857–1859, 2015.
[35]. K.-C. Chou, “Prediction of protein subcellular locations by incorporating quasi-sequence-order effect, ” Biochem. Biophys. Res. Commun., vol. 278, no. 2, pp. 477–483, 2000.
[36]. L. Wei, C. Zhou, H. Chen, J. Song, and R. Su, “ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides, ” Bioinformatics, vol. 34, no. 23, pp. 4007–4016, 2018.
[37]. T. Zhang, Y. Jia, H. Li, D. Xu, J. Zhou, and G. Wang, “CRISPRCasStack: A stacking strategy-based ensemble learning framework for accurate identification of Cas proteins, ” Brief. Bioinform., vol. 23, no. 5, p. bbac335, 2022.