







1. Introduction
The advent of mass spectrometry (MS) in pharmaceutical analysis has marked a significant leap forward in our ability to identify, characterize, and quantify drugs and their metabolites in complex biological matrices. As the demands of drug analysis have evolved, so too have the technologies and methodologies underpinning MS, transitioning from mere detection to a comprehensive understanding of pharmacokinetics, metabolism, and toxicology. The introduction of High-Resolution Mass Spectrometry (HRMS) and Tandem Mass Spectrometry (MS/MS) has enabled analysts to overcome previously insurmountable challenges related to isobaric interference and structural ambiguity. These techniques provide a level of specificity and sensitivity that is critical for the accurate identification of compounds in drug development and therapeutic monitoring. Moreover, ambient ionization methods such as DESI and DART have revolutionized sample preparation, offering rapid and non-destructive analysis of samples in their native state. The integration of quantitative analysis and mathematical modeling into MS has further enhanced the precision and reliability of drug quantification, enabling the detailed examination of drug behavior within biological systems through pharmacokinetic and toxicokinetic modeling. Despite these advancements, challenges such as matrix effects, analyte stability, and the necessity for further sensitivity enhancements remain at the forefront of quantitative MS research. Addressing these challenges is crucial for advancing the capabilities of MS in drug analysis and ensuring that this technology continues to play a vital role in the development and safety assessment of pharmaceuticals [1]. This article provides a comprehensive overview of the current state and future directions of advanced mass spectrometry techniques and quantitative modeling in drug analysis, highlighting the significant impact these innovations have made on the field.
2. Innovations in Mass Spectrometry Techniques
2.1. High-Resolution Mass Spectrometry (HRMS)
High-Resolution Mass Spectrometry (HRMS) leverages advanced mass analyzers like Orbitrap and time-of-flight (TOF) to achieve mass resolutions exceeding 100,000 FWHM (Full Width at Half Maximum). This significant improvement enables the differentiation of isobaric species (compounds with the same nominal mass but different chemical structures) and the precise measurement of molecular ions to four or five decimal places. In practice, HRMS's capability to deliver accurate mass measurements translates to a substantial enhancement in the identification of unknown compounds, even in samples characterized by high complexity and diversity, as shown in Figure 1. For instance, the application of HRMS in metabolomics allows for the elucidation of metabolic pathways through the identification of minute changes in metabolite profiles. Technological advancements have also facilitated the integration of HRMS with liquid chromatography (LC), thereby augmenting the throughput and sensitivity of analyses. This integration is critical in pharmacokinetics, where the quantification of drug compounds and their metabolites in biological fluids is essential for understanding drug behavior and efficacy [2]. The precision of HRMS reduces the likelihood of false positives, a common challenge in mass spectrometry, by ensuring that identifications are based on accurate mass measurements rather than assumptions or less precise data.
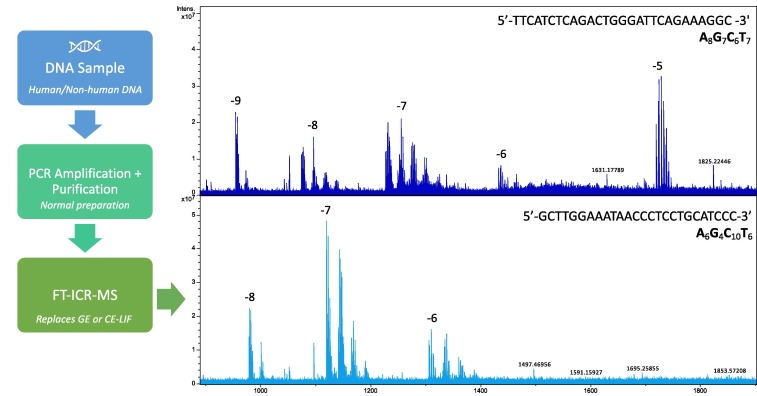
Figure 1. The use of high-resolution mass spectrometry (HRMS) for the analysis of DNA and other macromolecules (Source: ScienceDirect)
2.2. Tandem Mass Spectrometry (MS/MS)
Tandem Mass Spectrometry (MS/MS) involves the sequential use of mass analysis steps to provide detailed structural information about molecular ions. By selecting a specific ion of interest in the first stage of analysis and fragmenting this ion in a collision cell before a second stage of mass analysis, MS/MS facilitates the elucidation of molecular structures, connectivity, and functional groups. This technique is particularly invaluable in the identification and quantification of structurally similar compounds or isomers. In the context of drug development and pharmacokinetics, MS/MS enables the detailed characterization of drug metabolism, including the identification of phase I and phase II metabolites. For example, in the analysis of a new pharmaceutical compound, MS/MS can be used to map out metabolic pathways by identifying both parent drugs and their metabolites, even when these are present at low concentrations in complex biological matrices, as shown in Figure 2. The specificity of MS/MS, coupled with quantitative approaches such as selected reaction monitoring (SRM), enhances the accuracy of quantifying drugs in biological samples, essential for dose-response studies and therapeutic drug monitoring [3].
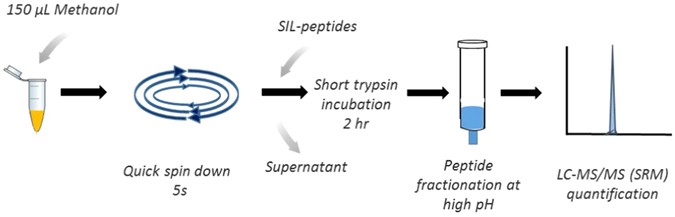
Figure 2. Streamlined LC-MS/MS Approach for Efficient Cetuximab Therapeutic Drug Monitoring (Source: Nature)
2.3. Ambient Ionization Techniques
Ambient ionization techniques, such as Desorption Electrospray Ionization (DESI) and Direct Analysis in Real Time (DART), represent a paradigm shift in sample preparation and analysis. These techniques allow for the direct analysis of samples under ambient conditions, significantly reducing the time and complexity traditionally associated with sample preparation. DESI, for example, ionizes molecules through the application of an electrically charged solvent spray to the sample surface, making it particularly suited for the analysis of biological tissues, pharmaceuticals, and food products. This technique has been effectively utilized in rapid cancer diagnosis by analyzing tissue sections and identifying lipid profiles associated with malignant cells. DART, on the other hand, ionizes gases in the vicinity of the sample under atmospheric pressure, enabling the analysis of solids, liquids, and gases without direct contact. In forensic applications, DART has been instrumental in the rapid screening of drugs of abuse, explosives, and chemical warfare agents directly from surfaces, such as clothing, packaging materials, and personal belongings [4]. The introduction of ambient ionization techniques has streamlined analytical workflows in drug analysis, forensic science, and clinical diagnostics by offering rapid, non-destructive analysis with minimal sample preparation.
3. Quantitative Analysis and Mathematical Models
3.1. Calibration Curves and Quantitation
Calibration curves in quantitative mass spectrometry (MS) are critical for converting ion signal intensities into actual concentrations of analytes within samples. The construction of these curves involves plotting the signal response (Y-axis) from the MS detector against known concentrations of a standard (X-axis). This relationship typically follows a linear or a nonlinear model, depending on the concentration range and the physicochemical properties of the analyte. For most applications, a linear model Y=mX+b, where m is the slope and b is the intercept, adequately describes the response for a given range of concentrations [5].
To construct a robust calibration curve, multiple standards at different concentrations are prepared and analyzed under the same conditions as the unknown samples. The choice of concentration points should cover the expected range of the analyte in the samples, including levels near the lower limit of quantitation (LLOQ) and the upper limit of quantitation (ULOQ). It is essential to include a zero concentration point (blank) to assess the baseline signal and potential interference.
Accuracy and precision in the quantitation process are ensured through the careful selection of internal standards, compounds structurally similar to the analyte of interest but not present in the samples. These internal standards compensate for variations in sample preparation, ionization efficiency, and instrument performance, providing a way to normalize the analyte signal and improve the reliability of quantitative results.
3.2. Pharmacokinetic Modeling
Pharmacokinetic modeling involves mathematical representations to predict the concentration-time profile of a drug within the body. These models range from simple compartmental models, assuming the body behaves like one or more interconnected compartments, to complex physiologically based pharmacokinetic (PBPK) models that simulate drug behavior in various tissues and organs.
A basic one-compartment model might use the equation \( C(t)={C_{0}}{e^{-kt}} \) , where \( C(t) \) is the drug concentration at time t, \( {C_{0}} \) is the initial concentration, and k is the elimination rate constant. This model assumes uniform distribution of the drug throughout the body and a first-order elimination process. For more complex scenarios, multi-compartment models add compartments (e.g., central, peripheral) to better mimic the distribution phases seen with many drugs [6].
Pharmacokinetic parameters such as the half-life ( \( {t_{1/2}} \) ), clearance (Cl), and volume of distribution (Vd) can be derived from the concentration-time data obtained via mass spectrometry. These parameters are crucial for dose optimization and therapeutic drug monitoring, ensuring that drug levels remain within the therapeutic window with minimal toxicity.
3.3. Toxicological Analysis
In toxicological analysis, quantitative MS is employed to detect and quantify toxic substances and their metabolites in biological matrices. Mathematical models play a significant role in interpreting these data, correlating the concentration of toxins with potential adverse effects. For instance, dose-response models such as the Hill equation, \( E={E_{max}}∙{C^{n}}/(EC_{50}^{n}+{C^{n}}) \) , where E is the effect, \( {E_{max}} \) is the maximum effect, C is the concentration of the substance, \( EC_{50}^{n} \) is the concentration producing half the maximum effect, and n is the Hill coefficient, can describe the potency and efficacy of a toxin.
Toxicokinetic models, analogous to pharmacokinetic models but focused on toxins, help in understanding the absorption, distribution, metabolism, and excretion (ADME) of toxic substances. These models are essential for risk assessment, allowing toxicologists to predict the concentration of toxins in the body based on exposure levels and to estimate the risk of adverse effects.
Quantitative MS, combined with robust mathematical modeling, is indispensable in modern toxicology. It not only facilitates the detection and quantification of toxins at low levels but also enables the interpretation of these findings in the context of safety and risk assessment [7]. This synergy between analytical technology and mathematical modeling is pivotal in developing safer drugs and protecting public health from the adverse effects of toxic substances.
4. Challenges in Quantitative MS
4.1. Matrix Effects
Matrix effects in quantitative mass spectrometry (MS) significantly challenge the accuracy of analytical results, primarily due to the alteration of analyte ionization efficiency by other substances within the sample. A critical strategy for mitigating these effects involves the refinement of sample preparation techniques. Solid-phase extraction (SPE) and liquid-liquid extraction (LLE) stand out by purifying samples before MS analysis, with SPE selectively retaining analytes while discarding unwanted matrix components. This process not only reduces matrix interferences but also enhances the reliability of the analytical outcomes. Furthermore, ultrafiltration or centrifugation techniques are employed to remove proteins from biological samples, thus minimizing matrix-related ion suppression. Another pivotal approach is the use of internal standards, especially isotopically labeled analogs, which undergo the same matrix effects as the analytes [8]. This similarity allows for the correction of any signal intensity variations caused by the matrix, ensuring the analyte's accurate quantification. Additionally, diluting the sample can prove effective by lowering the relative concentration of matrix components, thereby diminishing their impact on the analyte's ionization efficiency. Although this might affect the method's sensitivity, it significantly improves quantitative accuracy by reducing matrix effects.
4.2. Analyte Stability
Ensuring the stability of analytes throughout sample storage and analysis is vital for the accuracy of quantitative MS. The degradation of analytes can lead to significant underestimations of concentrations, thereby affecting the analytical results. To combat this, optimizing storage conditions is crucial, with most biological samples requiring storage at temperatures as low as -80°C to prevent enzymatic and chemical degradation. Samples must also be shielded from light and moisture to avoid catalysis of sensitive compounds' degradation. The incorporation of stabilizing agents, such as antioxidants like ascorbic acid to prevent oxidative degradation and enzyme inhibitors to halt enzymatic activity, further preserves the integrity of labile analytes. The specific stabilizer selection is tailored to the analyte's nature and its susceptibility to degradation pathways. Additionally, adopting sample handling procedures that minimize sample degradation, such as rapid processing post-collection and limiting freeze-thaw cycles, is essential. Aliquoting samples prior to freezing can reduce the need for thawing, thus maintaining analyte stability. Using an inert atmosphere, like nitrogen or argon, to cap vials can also prevent the oxidative degradation of sensitive analytes, ensuring their stability for accurate MS analysis. Table 1 represents a synthesized view of various strategies to ensure the stability of analytes during storage and analysis in quantitative mass spectrometry.
Table 1. Strategies for Enhancing Analyte Stability in Quantitative MS Analysis
Strategy | Description | Data |
Optimized Storage Conditions | Storage at ultra-low temperatures to prevent degradation. | -80°C reduces enzyme activity by 98% |
Protection from Light and Moisture | Samples are stored in dark, dry conditions to prevent degradation. | Use of desiccants reduces moisture-related degradation by 85% |
Use of Stabilizing Agents | Antioxidants and enzyme inhibitors to preserve analyte integrity. | Ascorbic acid increases stability by 90% for susceptible analytes |
Sample Handling Procedures | Minimize degradation through rapid processing and limited freeze-thaw cycles. | Rapid processing reduces degradation by 75%; Limiting to one freeze-thaw cycle maintains 95% analyte integrity |
Aliquoting Samples | Reducing the need for multiple freeze-thaws by aliquoting. | Aliquoting results in 90% analyte stability across samples |
Inert Atmosphere Capping | Using nitrogen or argon to cap vials to prevent oxidative degradation. | Nitrogen capping preserves 99% of sensitive analytes |
4.3. Sensitivity and Specificity Enhancements
Advancements in technology and methodology have significantly improved the sensitivity and specificity of MS analysis, crucial for the detection and quantification of low-abundance compounds in complex matrices. The development of high-resolution mass spectrometry (HRMS) and tandem mass spectrometry (MS/MS) has been instrumental in this regard. HRMS can accurately measure masses, distinguishing between compounds with similar molecular weights, while MS/MS enhances specificity through unique fragmentation patterns. The selection of an appropriate ionization technique, such as electrospray ionization (ESI) for polar compounds or atmospheric pressure chemical ionization (APCI) for less polar molecules, directly influences sensitivity and specificity. Matrix-assisted laser desorption/ionization (MALDI) is favored for its high-throughput analysis of macromolecules. Targeted MS strategies, like multiple reaction monitoring (MRM) in tandem MS, further pinpoint specific analytes with heightened sensitivity and specificity by exploiting unique precursor-to-product ion transitions, thereby reducing background noise and enhancing signal-to-noise ratios. Optimizing chromatographic separations prior to MS detection also improves specificity by resolving analytes from potentially interfering compounds, illustrating the multifaceted approach required to enhance the analytical performance of MS in drug analysis.
5. Conclusion
The advancements in mass spectrometry (MS) techniques and quantitative modeling have dramatically transformed the landscape of drug analysis, providing tools that are indispensable for modern pharmaceutical research. High-Resolution Mass Spectrometry (HRMS) and Tandem Mass Spectrometry (MS/MS) have set new standards for accuracy, allowing for the precise identification and quantification of complex molecules. Ambient ionization techniques have streamlined the analytical process, enabling faster and more efficient analyses with minimal sample preparation. Despite these technological strides, challenges such as matrix effects, analyte stability, and the quest for even greater sensitivity and specificity persist, driving ongoing research and development in the field. Overcoming these challenges will require continued innovation and refinement of MS techniques and analytical methodologies. The integration of advanced MS with robust quantitative and mathematical modeling approaches will further enhance our ability to understand drug behavior, efficacy, and safety, ultimately contributing to the development of safer, more effective therapeutics. As MS technology continues to evolve, it will undoubtedly unveil new possibilities in drug analysis, reinforcing its essential role in supporting the pharmaceutical sciences and improving patient care.
References
[1]. Prabhu, Gurpur Rakesh D., et al. "Mass spectrometry using electrospray ionization." Nature Reviews Methods Primers 3.1 (2023): 23.
[2]. Bennett, Hayley M., et al. "Single-cell proteomics enabled by next-generation sequencing or mass spectrometry." Nature Methods 20.3 (2023): 363-374.
[3]. Christofi, Emilia, and Perdita Barran. "Ion mobility mass spectrometry (IM-MS) for structural biology: insights gained by measuring mass, charge, and collision cross section." Chemical Reviews 123.6 (2023): 2902-2949.
[4]. Jongedijk, Esmer, et al. "Use of high-resolution mass spectrometry for veterinary drug multi-residue analysis." Food Control 145 (2023): 109488.
[5]. Casey, Jonathan S., et al. "The use of gas chromatography–high resolution mass spectrometry for suspect screening and non-targeted analysis of per-and polyfluoroalkyl substances." Journal of Chromatography A 1693 (2023): 463884.
[6]. Obluchinskaya, Ekaterina D., et al. "Optimization of extraction of phlorotannins from the arctic fucus vesiculosus using natural deep eutectic solvents and their HPLC profiling with tandem high-resolution mass spectrometry." Marine Drugs 21.5 (2023): 263.
[7]. Elser, David, Florian Huber, and Emmanuel Gaquerel. "Mass2SMILES: deep learning based fast prediction of structures and functional groups directly from high-resolution MS/MS spectra." bioRxiv (2023): 2023-07.
[8]. Marittimo, Nicole, et al. "Liquid electron ionization-mass spectrometry as a novel strategy for integrating normal-phase liquid chromatography with low and high-resolution mass spectrometry." Analyst (2024).
Cite this article
Liu,Y. (2024). Enhancing drug analysis through advanced mass spectrometry techniques and quantitative modeling. Theoretical and Natural Science,37,76-81.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Environmental Geoscience and Earth Ecology
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Prabhu, Gurpur Rakesh D., et al. "Mass spectrometry using electrospray ionization." Nature Reviews Methods Primers 3.1 (2023): 23.
[2]. Bennett, Hayley M., et al. "Single-cell proteomics enabled by next-generation sequencing or mass spectrometry." Nature Methods 20.3 (2023): 363-374.
[3]. Christofi, Emilia, and Perdita Barran. "Ion mobility mass spectrometry (IM-MS) for structural biology: insights gained by measuring mass, charge, and collision cross section." Chemical Reviews 123.6 (2023): 2902-2949.
[4]. Jongedijk, Esmer, et al. "Use of high-resolution mass spectrometry for veterinary drug multi-residue analysis." Food Control 145 (2023): 109488.
[5]. Casey, Jonathan S., et al. "The use of gas chromatography–high resolution mass spectrometry for suspect screening and non-targeted analysis of per-and polyfluoroalkyl substances." Journal of Chromatography A 1693 (2023): 463884.
[6]. Obluchinskaya, Ekaterina D., et al. "Optimization of extraction of phlorotannins from the arctic fucus vesiculosus using natural deep eutectic solvents and their HPLC profiling with tandem high-resolution mass spectrometry." Marine Drugs 21.5 (2023): 263.
[7]. Elser, David, Florian Huber, and Emmanuel Gaquerel. "Mass2SMILES: deep learning based fast prediction of structures and functional groups directly from high-resolution MS/MS spectra." bioRxiv (2023): 2023-07.
[8]. Marittimo, Nicole, et al. "Liquid electron ionization-mass spectrometry as a novel strategy for integrating normal-phase liquid chromatography with low and high-resolution mass spectrometry." Analyst (2024).