







1. Introduction
As a prevailing type of Temporal Lobe Epilepsy, Mesial Temporal Lobe Epilepsy happens mostly in the middle part of the temporal lobe, which involves the hippocampus and surrounding areas. Temporal Lobe epilepsy can be detected through MRI, where one can sometimes observe hippocampal sclerosis. It is also a widespread neuronal dysfunction in the central nervous system [1]. Additionally, several medications have already appeared to be efficient in treating mesial temporal lobe epilepsy, which includes Carbamazepine, Gabapentin, Lamotrigine, Topiramate, and Oxcarbazepine [2].
NKCC1, encoded by Slc12a2 and thought to be the cost of 30% of epilepsies, is a protein that is commonly found in neurons in the central nervous system, peripheral nervous system, and some glial cells and is responsible for the uptake of chloride ions from the extracellular matrix. NKCC1 predominates in the immature neuron, while KCC2 is kept in the minority due to some age-dependent factor, causing the chloride concentration higher within the cell, making the neuron excitatory. However, as the neuron matures, KCC2 upgrades and NKCC1 degrades, making the chloride concentration within and without cell alters, turning the neuron to be inhibitory [3].
Although not yet determined, the cause at a molecular level for mesial temporal lobe epilepsy had long been a prevailing topic, in this article, we focused on NKCC1 as a potential cause for this disease and try to set up a corresponding theoretical framework that justifies NKCC1 and its associated mechanism as a potential cause for mesial temporal lobe epilepsy, and will provide expectations for medicine accordingly.
2. Current Understanding Of The Potential Causes For Mesial Temporal Lobe Epilepsy
Taking up about 80% of all epilepsy, the cause of mesial temporal lobe epilepsy (MTLE) at a micro level remains unknown, it is often observed many macro or general causes would induce mesial temporal lobe epilepsy, which may include physical traumas in the brain area, genetic inheritances, and Neurocysticercosis according to a recently published paper [4]. Therefore, throughout the decades, researchers have hypothesized and testified several possible causes.
2.1. Reelin loss
There are varied explanations for the possible causes of MTLE, one of them that had been convincing in the decades is the theory about the loss of Reelin, a secreted neurodevelopmental glycoprotein, has long been thought to be associated with the formation of MTLE. In a paper in 2006[4], they proposed that loss of Reelin in the epileptic adult hippocampus induced aberrant integration of newborn Granule Cell Dispersions (GDC) [4]. Then later in a paper published in 2022, [5], they provided insight into the effect of Reelin, GDCs on mesial temporal lobe epilepsy, whereas they suspected that loss of Reelin affected GDCs, which is commonly found in epileptic brains [5]. This suspicion is then being supported by research in 2023 [6]. In this research, they found that loss of Reelin induced a decrease in its downstream target, disabled 1 (Dab1), which inhibits cofilin to avoid aberrant neuronal migration. Moreover, they found in the patient a significant overexpression of Cofilin, indicating the pathway they introduced to be quite plausible [6].
2.2. miRNA-induced epilepsy.
miRNA, as to Reelin, has been suspected for an extended period to be one of the causes of epilepsy. In a paper published in 2011 [7]. They first demonstrated a miRNA alternation after seizures [7]. Later, as more studies about miRNA’s role in Temporal Lobe Epilepsy, the idea that miRNA affected some pathological responses in the brain area eventually led to epilepsy [8]. In a study in 2016 [9], they suggested that a decrease of generalized miR-146a-mediated leads to a decrease of complement factor H and is likely to induce temporal lobe epilepsy in the rat model [9]. Moreover, other studies suggested that miRNA is associated with neural function and plasticity that may also contribute as a factor for temporal lobe epilepsy [8]. For instance, in research in 2013 [10], the research group demonstrated that miRNA expression alternation induced functional changes in the dentate gyrus and thus had a large impact on causing temporal lobe epilepsy [10].
2.3. Blood-brain barrier (BBB) leakage induces temporal lobe epilepsy.
Unlike Reelin losses and miRNA dysregulation, Blood-Brain Barrier disruption is a rather new and minor area of consideration on what induced temporal lobe epilepsy. However, immune cells’ transmigration or invasion into the brain parenchyma can also induce epilepsy [11]. This had been supported by a paper published in 2008 [12]. They demonstrated a pathogenetic link between leukocyte-vascular interaction, BBB damage, and the formation of seizures [12].
However, in a recently published paper in 2022[11], the research group applied functional magnetic resonance imaging (fMRI) to 90 subjects with Temporal Lobe Epilepsy (TLE) and ran a statistical experiment to test the patients’ functional connectivity in their brain areas. They demonstrated that the memory network of the TLE patients had experienced changes inside the mesial temporal lobe and frontal lobe when compared to the control group, and a higher level of disease burden is associated with weaker connectivity with the inter-mesial temporal lobe and intra-mesial temporal lobe, which support verbal and visual memory [11]. These new findings inspired a new possible mechanism for explaining the fundamental cause: the role of hippocampal pyramidal cells in causing mesial temporal lobe epilepsy.
3. Nkcc1 Upregulation In The Hippocampal Cells Induced Mesial Temporal Lobe Epilepsy
Hippocampal pyramidal cells are learned to be associated with spatial learning [13]. Thus, functional disability of hippocampal pyramidal cells would cause alternations in spatial learning and furthermore affects episodic memory. Moreover, hippocampal pyramidal cells contain NKCC1 and KCC2 proteins [14], which are recently found to be key proteins in inducing epilepsy [15]. Therefore, we hypothesized such following mechanism: upregulation of NKCC1 associated with downregulation of KCC2 [16] induced intracellular chloride ions accumulation, thus, alternating the charge within the neurons turning it from inhibitory to excitatory [15], causing hyperexcitement, which damaged the original function of hippocampal pyramidal cells, causes epilepsies, and disrupts episodic memory. In some cases, a high level of intracellular chloride ions would become toxic for the neurons, therefore inducing more than 50% of cells in the hippocampus to go through apoptosis and inflammation, which on a macro scale would display as hippocampal sclerosis [17]. Additionally, this hypothesized mechanism follows the pattern of neural maturation. Since NKCC1 must experience a downregulation while KCC2 has to experience an upregulation as the neuron matures, any mutation or functional disability during this process of transforming would result in an outbalanced NKCC1 and KCC2 ratio, which makes the maturing stage of the neurons having the most threats for experiencing hyperexcitement. This provides a plausible explanation for why it is common for the mesial temporal lobe to display epilepsy since it is one of the areas that mature first in the brain [18].
3.1. WNK-SPAK/OSR1 function in regulating NKCC1 and KCC2 and as possible cause for epilepsy
Several recently published papers have extended our previous hypothesis and have adjusted our focus on the whole mechanism. In these two papers [19] [20], WNK had been demonstrated as both chloride concentration gated, and cell volume gated (Figure), whereas the cell is exposed to a hypertonic Cl environment, the cell would go through hyperosmotic.
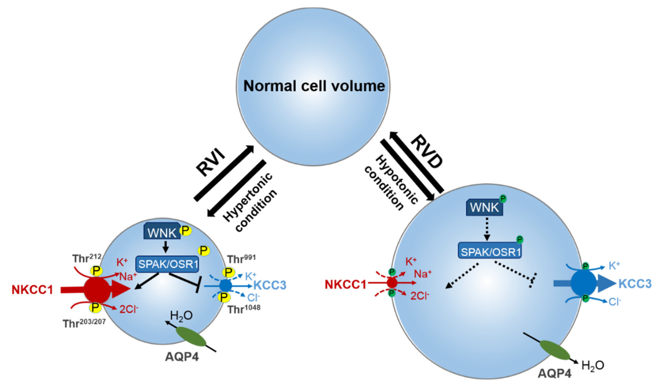
Figure 1. Roles of WNK, NKCC1, and KCC2 on regulating cell volume and intracellular ion concentration [20].
Thus, activating the WNK-SPAK/OSR1, and upregulating NKCC1 to bring in Cl to restore cell volume. Moreover, the WNK pathway had been found to regulate NKCC1 and KCC2 at the same time, and when WNK is activated, it upregulates NKCC1 as well as downregulates KCC2[19] [20]. And as research indicated that WNK has an autophosphorylation mechanism [21], it remained unclear how WNK function in immature neurons. According to existing researchers, in immature neurons where intracellular chloride is much higher than in mature neurons, the WNK pathway should be inhibited, and NKCC1 should be downregulated while KCC2 should be upregulated, but NKCC1 still prevails in immature neurons. There must present an unknown factor that regulates WNK and he co-transporters. Though some mechanisms had been found to furthermore regulate WNK, NKCC1, and KCC2 (Figure 2), more are yet to be discovered, since these factors, when malfunctioning or mutated, have the potential to disrupt the expression of WNK or directly disrupt the co-transporters, thus inducing hyperexcitement and causing epilepsy. Therefore, it is critical to identify as many as mechanisms to avoid many unknown causes of epilepsies as possible.
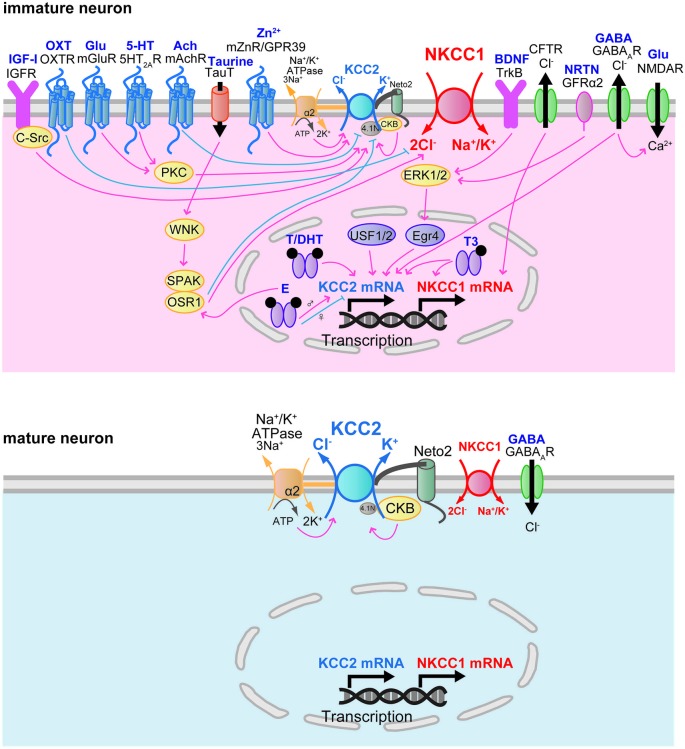
Figure 2. Currently known proteins for regulating WNK, NKCC1, and KCC2[18].
3.2. Possible mechanisms other than the WNK pathway in regulating NKCC1 and KCC2.
We further categorized the mechanisms that regulate WNK, NKCC1, and KCC2 into three categories: an unknown mechanism that regulates WNK, an unknown mechanism that regulates KCC2/NKCC1 along with WNK, different regulation patterns of WNK in immature and mature neurons (Figure3). And all three these categories do not stand against each other, they can be stand-alone, dependent on each other, and even coexist.
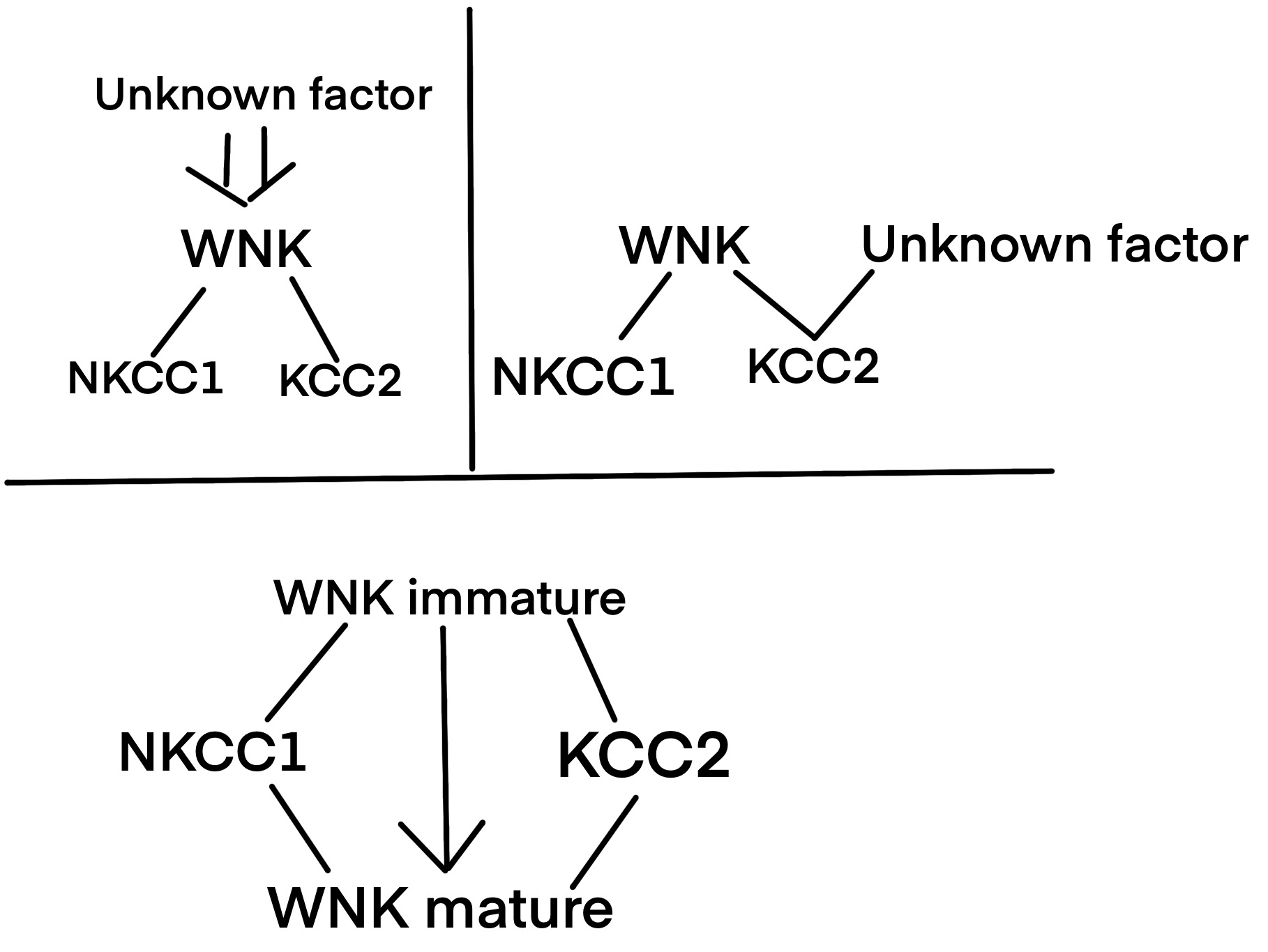
Figure 3. Three possible categories of mechanisms regulate WNK, NKCC1, and KCC2.
For the first categories, the unknown factor regulates WNK and furthermore regulates the co-transporters, has examples of Kelch-like 3(KLHL3) and Cullin-Ring ubiquitin 3 (CUL3) [22], which both would inhibit WNK kinase expression. KLHL3 BTB binds the C-terminus of L-WNK1 or WNK4, and Ring-box protein 1 of CUL3 binds to KLHL3 in the BTB domain, using enzymes that would degrade the two WNKs, and thus is inhibited [22]. Other than KLHLU3 and CUL3, Taurine happens to be another natural-occurring inhibitor or WNK family [18]. Another type of WNK regulator is SGK1, but SGK1 and WNK are in bilateral regulation (specifically for WNK1 and WNK4), whereas SGK1 phosphorylates WNK at S1169 and S1196[22], while the WNK family’s N termini displayed effectiveness to strong activation of SGK1 [23].
In the second category that WNK and an unknown factor both regulate the co-transporters, a paper published in 2017 provided an example that when spinal alpha-7 nicotinic acetylcholine receptor (nAChR) is activated, it upregulates KCC2 in rats [24], whereas brain-derived neurotrophic factor/tyrosine receptor kinase B expressions are reduced by nAChR that leads to an upregulation of KCC2. Moreover, in a paper published in 2019, during Hypoxic-ischemic encephalopathy (HIE), hypoxia-inducible factor-1α (HIF-1α) upregulates NKCC1 and nuclear factor of activated T cells 5 (NFAT5) that down-regulates NKCC1 [25]. This mechanism serves another typical type of multiple factors regulation on the cotransporters.
The last category of the mechanism, the difference in functions in mature and immature central nervous systems, has a limited number of studies compared to the other two categories. However, there is still insight into this category. In a paper published in 2021, WNKs are potassium-sensitive kinases [26], proposed that potassium is a chloride-independent WNK inhibitor. Moreover, the paper demonstrated a potassium secretion increase when bathed in a hypotonic environment, following a WNK-, Fray-, NKCC- dependent manner [26]. This provides insight into a possible mechanism of WNK in an immature state of neurons, where we can hypothesize that WNK in immature neurons is more potassium-gated than chloride-gated.
4. Envisioned Treatment (Medication)
For mesial temporal lobe epilepsy induced by NKCC1 overexpression, we do not believe medications should be inhibiting NKCC1 itself, even though it’s the direct cause of the disease. In most cases, the upregulation of NKCC1 often comes with the downregulation of KCC2 [19] [20], which makes the cell vulnerable to hypertonic environments and increases the percentage of apoptosis in such an environment. Thus, if a medication inhibits NKCC1 protein, it may release the symptom for a limited period, but it potentially decreases the cell’s ability to respond to hypotonic environments as well, which would eventually proliferate the threat on cell apoptosis. Under such saying, we think the medication should be targeting the WNK pathway or other mechanism that regulates the co-transporters, wherein such means, we could restore the organism’s ability for maintaining homeostasis, and thus self-cure epilepsy.
5. Conclusion
Through the analysis of the current understanding of mesial temporal lobe epilepsy, this article introduced several possible causes of mesial temporal lobe epilepsy and focused on one cause to analyze its current stage and propose future development. The article also includes envisioning medications for the specific cause of the disease previously described. The causes of epilepsy are diverse and largely varied, and the process of uncovering the causes at a molecular level could be time-taking and burdening. However, finding the cause of a disease at a molecular level means the discovery of potential targets, and potential biomarkers, which leads to a higher cure rate and early detection. Under such a saying, it has always been critical to seek the potential cause of every disease.
References
[1]. Cascino GD. Widespread Neuronal Dysfunction in Temporal Lobe Epilepsy. Epilepsy Curr. 2003 Jan;3(1):31-32. doi: 10.1111/j.1535-7597.2003.03113.x. PMID: 15309105; PMCID: PMC321161.
[2]. Marson AG, Al-Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, Cramp C, Cockerell OC, Cooper PN, Doughty J, Eaton B, Gamble C, Goulding PJ, Howell SJ, Hughes A, Jackson M, Jacoby A, Kellett M, Lawson GR, Leach JP, Nicolaides P, Roberts R, Shackley P, Shen J, Smith DF, Smith PE, Smith CT, Vanoli A, Williamson PR; SANAD Study group. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet. 2007 Mar 24;369(9566):1000-15. doi: 10.1016/S0140-6736(07)60460-7. PMID: 17382827; PMCID: PMC2080688.
[3]. Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J Neurobiol. (1997) 33:781–95. 10.1002/(SICI)1097-4695(19971120)33:6<781::AID-NEU6>3.0.CO;2-5
[4]. Secchi TL, Brondani R, Bragatti JA, Bizzi JWJ, Bianchin MM. Evaluating the Association of Calcified Neurocysticercosis and Mesial Temporal Lobe Epilepsy With Hippocampal Sclerosis in a Large Cohort of Patients With Epilepsy. Front Neurol. 2022 Jan 27;12:769356. doi: 10.3389/fneur.2021.769356. PMID: 35153977; PMCID: PMC8830344.
[5]. Gong C, Wang TW, Huang HS, Parent JM. Reelin regulates neuronal progenitor migration in the intact and epileptic hippocampus. J Neurosci. 2007 Feb 21;27(8):1803-11. doi: 10.1523/JNEUROSCI.3111-06.2007. PMID: 17314278; PMCID: PMC6673551.
[6]. Leifeld J, Förster E, Reiss G, Hamad MIK. Considering the Role of Extracellular Matrix Molecules, in Particular Reelin, in Granule Cell Dispersion Related to Temporal Lobe Epilepsy. Front Cell Dev Biol. 2022 Jun 6;10:917575. doi: 10.3389/fcell.2022.917575. PMID: 35733853; PMCID: PMC9207388.
[7]. Gupta T, Kaur M, Singla N, Radotra BD, Sahni D, Kharbanda PS, Gupta SK. Reelin Signaling Pathway and Mesial Temporal Lobe Epilepsy: A Causative Link. Basic Clin Neurosci. 2023 Jan-Feb;14(1):57-72. doi: 10.32598/bcn.2021.2554.1. Epub 2023 Jan 1. PMID: 37346868; PMCID: PMC10279991.
[8]. Nudelman AS, DiRocco DP, Lambert TJ, Garelick MG, Le J, Nathanson NM, Storm DR. Neuronal activity rapidly induces transcription of the CREB-regulated microRNA-132, in vivo. Hippocampus. 2010 Apr;20(4):492-8. doi: 10.1002/hipo.20646. PMID: 19557767; PMCID: PMC2847008.
[9]. Manna I, Fortunato F, De Benedittis S, Sammarra I, Bertoli G, Labate A, Gambardella A. Non-Coding RNAs: New Biomarkers and Therapeutic Targets for Temporal Lobe Epilepsy. Int J Mol Sci. 2022 Mar 11;23(6):3063. doi: 10.3390/ijms23063063. PMID: 35328484; PMCID: PMC8954985.
[10]. He F, Liu B, Meng Q, Sun Y, Wang W, Wang C. Modulation of miR-146a/complement factor H-mediated inflammatory responses in a rat model of temporal lobe epilepsy. Biosci Rep. 2016 Dec 23;36(6):e00433. doi: 10.1042/BSR20160290. PMID: 27852797; PMCID: PMC5180253.
[11]. Bot AM, Dębski KJ, Lukasiuk K. Alterations in miRNA levels in the dentate gyrus in epileptic rats. PLoS One. 2013 Oct 11;8(10):e76051. doi: 10.1371/journal.pone.0076051. PMID: 24146813; PMCID: PMC3795667.
[12]. Reiss Y, Bauer S, David B, Devraj K, Fidan E, Hattingen E, Liebner S, Melzer N, Meuth SG, Rosenow F, Rüber T, Willems LM, Plate KH. The neurovasculature as a target in temporal lobe epilepsy. Brain Pathol. 2023 Mar;33(2):e13147. doi: 10.1111/bpa.13147. Epub 2023 Jan 4. PMID: 36599709; PMCID: PMC10041171.
[13]. Fleury M, Buck S, Binding LP, Caciagli L, Vos SB, Winston GP, Thompson PJ, Koepp MJ, Duncan JS, Sidhu MK. Episodic memory network connectivity in temporal lobe epilepsy. Epilepsia. 2022 Oct;63(10):2597-2622. doi: 10.1111/epi.17370. Epub 2022 Aug 2. PMID: 35848050; PMCID: PMC9804196.
[14]. O’Keefe J, Krupic J. Do hippocampal pyramidal cells respond to nonspatial stimuli? Physiol Rev. 2021 Jul 1;101(3):1427-1456. doi: 10.1152/physrev.00014.2020. Epub 2021 Feb 16. PMID: 33591856; PMCID: PMC8490123.
[15]. Kurki SN, Uvarov P, Pospelov AS, Trontti K, Hübner AK, Srinivasan R, Watanabe M, Hovatta I, Hübner CA, Kaila K, Virtanen MA. Expression patterns of NKCC1 in neurons and non-neuronal cells during cortico-hippocampal development. Cereb Cortex. 2023 May 9;33(10):5906-5923. doi: 10.1093/cercor/bhac470. PMID: 36573432; PMCID: PMC10183754.
[16]. Liu R, Wang J, Liang S, Zhang G, Yang X. Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front Neurol. 2020 Jan 17;10:1407. doi: 10.3389/fneur.2019.01407. PMID: 32010056; PMCID: PMC6978738.
[17]. Côme E, Blachier S, Gouhier J, Russeau M, Lévi S. Lateral Diffusion of NKCC1 Contributes to Chloride Homeostasis in Neurons and Is Rapidly Regulated by the WNK Signaling Pathway. Cells. 2023 Jan 31;12(3):464. doi: 10.3390/cells12030464. PMID: 36766805; PMCID: PMC9914440.
[18]. Author links open overlay panelAndrew E. Budson M.D. (2015, July 7). Other disorders that cause memory loss or dementia. Memory Loss, Alzheimer’s Disease, and Dementia (Second Edition). https://www.sciencedirect.com/science/article/abs/pii/B9780323286619000147
[19]. Watanabe M, Fukuda A. Development and regulation of chloride homeostasis in the central nervous system. Front Cell Neurosci. 2015 Sep 24;9:371. doi: 10.3389/fncel.2015.00371. PMID: 26441542; PMCID: PMC4585146.
[20]. Côme E, Blachier S, Gouhier J, Russeau M, Lévi S. Lateral Diffusion of NKCC1 Contributes to Chloride Homeostasis in Neurons and Is Rapidly Regulated by the WNK Signaling Pathway. Cells. 2023 Jan 31;12(3):464. doi: 10.3390/cells12030464. PMID: 36766805; PMCID: PMC9914440.
[21]. Josiah, S. S., Meor Azlan, N. F., & Zhang, J. (2021, January 27). Targeting the WNK-SPAK/OSR1 pathway and cation-chloride cotransporters for the therapy of stroke. MDPI. https://www.mdpi.com/1422-0067/22/3/1232
[22]. Sun Q, Wu Y, Jonusaite S, Pleinis JM, Humphreys JM, He H, Schellinger JN, Akella R, Stenesen D, Krämer H, Goldsmith EJ, Rodan AR. Intracellular Chloride and Scaffold Protein Mo25 Cooperatively Regulate Transepithelial Ion Transport through WNK Signaling in the Malpighian Tubule. J Am Soc Nephrol. 2018 May;29(5):1449-1461. doi: 10.1681/ASN.2017101091. Epub 2018 Mar 30. PMID: 29602832; PMCID: PMC5967776.
[23]. Lin SC, Ma C, Chang KJ, Cheong HP, Lee MC, Lan YT, Wang CY, Chiou SH, Huo TI, Hsu TK, Tsai PH, Yang YP. The Post-Translational Modification Networking in WNK-Centric Hypertension Regulation and Electrolyte Homeostasis. Biomedicines. 2022 Sep 2;10(9):2169. doi: 10.3390/biomedicines10092169. PMID: 36140271; PMCID: PMC9496095.
[24]. Heise CJ, Xu BE, Deaton SL, Cha SK, Cheng CJ, Earnest S, Sengupta S, Juang YC, Stippec S, Xu Y, Zhao Y, Huang CL, Cobb MH. Serum and glucocorticoid-induced kinase (SGK) 1 and the epithelial sodium channel are regulated by multiple with no lysine (WNK) family members. J Biol Chem. 2010 Aug 13;285(33):25161-7. doi: 10.1074/jbc.M110.103432. Epub 2010 Jun 4. PMID: 20525693; PMCID: PMC2919078.
[25]. Yang XL, Zeng ML, Shao L, Jiang GT, Cheng JJ, Chen TX, Han S, Yin J, Liu WH, He XH, Peng BW. NFAT5 and HIF-1α Coordinate to Regulate NKCC1 Expression in Hippocampal Neurons After Hypoxia-Ischemia. Front Cell Dev Biol. 2019 Dec 13;7:339. doi: 10.3389/fcell.2019.00339. PMID: 31921851; PMCID: PMC6923656.
[26]. Gu W, Zhang W, Lei Y, Cui Y, Chu S, Gu X, Ma Z. Activation of spinal alpha-7 nicotinic acetylcholine receptor shortens the duration of remifentanil-induced postoperative hyperalgesia by upregulating KCC2 in the spinal dorsal horn in rats. Mol Pain. 2017 Jan-Dec;13:1744806917704769. doi: 10.1177/1744806917704769. PMID: 28425312; PMCID: PMC6997724.
Cite this article
Li,Z. (2024). Potential regulatory mechanisms for NKCC1 and KCC2 that induce temporal lobe epilepsy. Theoretical and Natural Science,46,65-71.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Cascino GD. Widespread Neuronal Dysfunction in Temporal Lobe Epilepsy. Epilepsy Curr. 2003 Jan;3(1):31-32. doi: 10.1111/j.1535-7597.2003.03113.x. PMID: 15309105; PMCID: PMC321161.
[2]. Marson AG, Al-Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, Cramp C, Cockerell OC, Cooper PN, Doughty J, Eaton B, Gamble C, Goulding PJ, Howell SJ, Hughes A, Jackson M, Jacoby A, Kellett M, Lawson GR, Leach JP, Nicolaides P, Roberts R, Shackley P, Shen J, Smith DF, Smith PE, Smith CT, Vanoli A, Williamson PR; SANAD Study group. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet. 2007 Mar 24;369(9566):1000-15. doi: 10.1016/S0140-6736(07)60460-7. PMID: 17382827; PMCID: PMC2080688.
[3]. Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J Neurobiol. (1997) 33:781–95. 10.1002/(SICI)1097-4695(19971120)33:6<781::AID-NEU6>3.0.CO;2-5
[4]. Secchi TL, Brondani R, Bragatti JA, Bizzi JWJ, Bianchin MM. Evaluating the Association of Calcified Neurocysticercosis and Mesial Temporal Lobe Epilepsy With Hippocampal Sclerosis in a Large Cohort of Patients With Epilepsy. Front Neurol. 2022 Jan 27;12:769356. doi: 10.3389/fneur.2021.769356. PMID: 35153977; PMCID: PMC8830344.
[5]. Gong C, Wang TW, Huang HS, Parent JM. Reelin regulates neuronal progenitor migration in the intact and epileptic hippocampus. J Neurosci. 2007 Feb 21;27(8):1803-11. doi: 10.1523/JNEUROSCI.3111-06.2007. PMID: 17314278; PMCID: PMC6673551.
[6]. Leifeld J, Förster E, Reiss G, Hamad MIK. Considering the Role of Extracellular Matrix Molecules, in Particular Reelin, in Granule Cell Dispersion Related to Temporal Lobe Epilepsy. Front Cell Dev Biol. 2022 Jun 6;10:917575. doi: 10.3389/fcell.2022.917575. PMID: 35733853; PMCID: PMC9207388.
[7]. Gupta T, Kaur M, Singla N, Radotra BD, Sahni D, Kharbanda PS, Gupta SK. Reelin Signaling Pathway and Mesial Temporal Lobe Epilepsy: A Causative Link. Basic Clin Neurosci. 2023 Jan-Feb;14(1):57-72. doi: 10.32598/bcn.2021.2554.1. Epub 2023 Jan 1. PMID: 37346868; PMCID: PMC10279991.
[8]. Nudelman AS, DiRocco DP, Lambert TJ, Garelick MG, Le J, Nathanson NM, Storm DR. Neuronal activity rapidly induces transcription of the CREB-regulated microRNA-132, in vivo. Hippocampus. 2010 Apr;20(4):492-8. doi: 10.1002/hipo.20646. PMID: 19557767; PMCID: PMC2847008.
[9]. Manna I, Fortunato F, De Benedittis S, Sammarra I, Bertoli G, Labate A, Gambardella A. Non-Coding RNAs: New Biomarkers and Therapeutic Targets for Temporal Lobe Epilepsy. Int J Mol Sci. 2022 Mar 11;23(6):3063. doi: 10.3390/ijms23063063. PMID: 35328484; PMCID: PMC8954985.
[10]. He F, Liu B, Meng Q, Sun Y, Wang W, Wang C. Modulation of miR-146a/complement factor H-mediated inflammatory responses in a rat model of temporal lobe epilepsy. Biosci Rep. 2016 Dec 23;36(6):e00433. doi: 10.1042/BSR20160290. PMID: 27852797; PMCID: PMC5180253.
[11]. Bot AM, Dębski KJ, Lukasiuk K. Alterations in miRNA levels in the dentate gyrus in epileptic rats. PLoS One. 2013 Oct 11;8(10):e76051. doi: 10.1371/journal.pone.0076051. PMID: 24146813; PMCID: PMC3795667.
[12]. Reiss Y, Bauer S, David B, Devraj K, Fidan E, Hattingen E, Liebner S, Melzer N, Meuth SG, Rosenow F, Rüber T, Willems LM, Plate KH. The neurovasculature as a target in temporal lobe epilepsy. Brain Pathol. 2023 Mar;33(2):e13147. doi: 10.1111/bpa.13147. Epub 2023 Jan 4. PMID: 36599709; PMCID: PMC10041171.
[13]. Fleury M, Buck S, Binding LP, Caciagli L, Vos SB, Winston GP, Thompson PJ, Koepp MJ, Duncan JS, Sidhu MK. Episodic memory network connectivity in temporal lobe epilepsy. Epilepsia. 2022 Oct;63(10):2597-2622. doi: 10.1111/epi.17370. Epub 2022 Aug 2. PMID: 35848050; PMCID: PMC9804196.
[14]. O’Keefe J, Krupic J. Do hippocampal pyramidal cells respond to nonspatial stimuli? Physiol Rev. 2021 Jul 1;101(3):1427-1456. doi: 10.1152/physrev.00014.2020. Epub 2021 Feb 16. PMID: 33591856; PMCID: PMC8490123.
[15]. Kurki SN, Uvarov P, Pospelov AS, Trontti K, Hübner AK, Srinivasan R, Watanabe M, Hovatta I, Hübner CA, Kaila K, Virtanen MA. Expression patterns of NKCC1 in neurons and non-neuronal cells during cortico-hippocampal development. Cereb Cortex. 2023 May 9;33(10):5906-5923. doi: 10.1093/cercor/bhac470. PMID: 36573432; PMCID: PMC10183754.
[16]. Liu R, Wang J, Liang S, Zhang G, Yang X. Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front Neurol. 2020 Jan 17;10:1407. doi: 10.3389/fneur.2019.01407. PMID: 32010056; PMCID: PMC6978738.
[17]. Côme E, Blachier S, Gouhier J, Russeau M, Lévi S. Lateral Diffusion of NKCC1 Contributes to Chloride Homeostasis in Neurons and Is Rapidly Regulated by the WNK Signaling Pathway. Cells. 2023 Jan 31;12(3):464. doi: 10.3390/cells12030464. PMID: 36766805; PMCID: PMC9914440.
[18]. Author links open overlay panelAndrew E. Budson M.D. (2015, July 7). Other disorders that cause memory loss or dementia. Memory Loss, Alzheimer’s Disease, and Dementia (Second Edition). https://www.sciencedirect.com/science/article/abs/pii/B9780323286619000147
[19]. Watanabe M, Fukuda A. Development and regulation of chloride homeostasis in the central nervous system. Front Cell Neurosci. 2015 Sep 24;9:371. doi: 10.3389/fncel.2015.00371. PMID: 26441542; PMCID: PMC4585146.
[20]. Côme E, Blachier S, Gouhier J, Russeau M, Lévi S. Lateral Diffusion of NKCC1 Contributes to Chloride Homeostasis in Neurons and Is Rapidly Regulated by the WNK Signaling Pathway. Cells. 2023 Jan 31;12(3):464. doi: 10.3390/cells12030464. PMID: 36766805; PMCID: PMC9914440.
[21]. Josiah, S. S., Meor Azlan, N. F., & Zhang, J. (2021, January 27). Targeting the WNK-SPAK/OSR1 pathway and cation-chloride cotransporters for the therapy of stroke. MDPI. https://www.mdpi.com/1422-0067/22/3/1232
[22]. Sun Q, Wu Y, Jonusaite S, Pleinis JM, Humphreys JM, He H, Schellinger JN, Akella R, Stenesen D, Krämer H, Goldsmith EJ, Rodan AR. Intracellular Chloride and Scaffold Protein Mo25 Cooperatively Regulate Transepithelial Ion Transport through WNK Signaling in the Malpighian Tubule. J Am Soc Nephrol. 2018 May;29(5):1449-1461. doi: 10.1681/ASN.2017101091. Epub 2018 Mar 30. PMID: 29602832; PMCID: PMC5967776.
[23]. Lin SC, Ma C, Chang KJ, Cheong HP, Lee MC, Lan YT, Wang CY, Chiou SH, Huo TI, Hsu TK, Tsai PH, Yang YP. The Post-Translational Modification Networking in WNK-Centric Hypertension Regulation and Electrolyte Homeostasis. Biomedicines. 2022 Sep 2;10(9):2169. doi: 10.3390/biomedicines10092169. PMID: 36140271; PMCID: PMC9496095.
[24]. Heise CJ, Xu BE, Deaton SL, Cha SK, Cheng CJ, Earnest S, Sengupta S, Juang YC, Stippec S, Xu Y, Zhao Y, Huang CL, Cobb MH. Serum and glucocorticoid-induced kinase (SGK) 1 and the epithelial sodium channel are regulated by multiple with no lysine (WNK) family members. J Biol Chem. 2010 Aug 13;285(33):25161-7. doi: 10.1074/jbc.M110.103432. Epub 2010 Jun 4. PMID: 20525693; PMCID: PMC2919078.
[25]. Yang XL, Zeng ML, Shao L, Jiang GT, Cheng JJ, Chen TX, Han S, Yin J, Liu WH, He XH, Peng BW. NFAT5 and HIF-1α Coordinate to Regulate NKCC1 Expression in Hippocampal Neurons After Hypoxia-Ischemia. Front Cell Dev Biol. 2019 Dec 13;7:339. doi: 10.3389/fcell.2019.00339. PMID: 31921851; PMCID: PMC6923656.
[26]. Gu W, Zhang W, Lei Y, Cui Y, Chu S, Gu X, Ma Z. Activation of spinal alpha-7 nicotinic acetylcholine receptor shortens the duration of remifentanil-induced postoperative hyperalgesia by upregulating KCC2 in the spinal dorsal horn in rats. Mol Pain. 2017 Jan-Dec;13:1744806917704769. doi: 10.1177/1744806917704769. PMID: 28425312; PMCID: PMC6997724.