







1. Introduction
Alzheimer’s disease (AD) is recognized as a complex and multifaceted neurological disorder, predominantly affecting the elderly population with a significant upsurge in incidence among individuals exceeding 65 years of age. Amidst the global trend of extended life expectancy, AD is anticipated to rise to the forefront as a primary cause of morbidity and mortality in the aging demographic, carrying substantial socioeconomic implications for society at large [1]. A wealth of scientific inquiry has confirmed the elevated levels of Aβ within the cerebral tissue of individuals afflicted with AD, where it accumulates in the form of extracellular deposits known as plaques. The aggregation of these senile plaques, alongside the apoptosis of neuronal cells instigated by Aβ, is identified as a pivotal contributor to the onset and progression of AD. Despite these insights, the existing methods for the detection of Aβ remain inadequate, and the challenge of quantifying Aβ with heightened sensitivity and precision persists.
AD’s complexity is further underscored by its multifaceted pathophysiology, encompassing neurodegeneration, Aβ accumulation, aberrant Tau phosphorylation, neuroinflammatory processes, and disruptions in glucose metabolism. Prior investigations have yet to fully decipher the intricate interplay among these pathological elements and their correlation to disease development. Considering these challenges, the present study aims to harness the potential of deep learning-driven protein structure prediction methodologies, encompassing advanced machine learning and artificial intelligence techniques, to mine large-scale datasets for concealed patterns and associations. This approach is poised to enhance our comprehension of the mechanisms underlying β-amyloid formation and its repercussions on the disease landscape.
2. Methods
2.1. Dataset
We obtained the ID of Aβ from the UniProt database: P05067-1 [2]. Its full sequence is: MLPGLALLLLAAWTARALEVPTDGNAGLLAEPQIAMFCGRLNMHMNVQNGKWDSDPSGTKTCIDTKEGILQYCQEVYPELQITNVVEANQPVTIQNWCKRGRKQCKTHPHFVIPYRCLVGEFVSDALLVPDKCKFLHQERMDVCETHLHWHTVAKETCSEKSTNLHDYGMLLPCGIDKFRGVEFVCCPLAEESDNVDSADAEEDDSDVWWGGADTDYADGSEDKVVEVAEEEEVAEVEEEEADDDEDDEDGDEVEEEAEEPYEEATERTTSIATTTTTTTESVEEVVREVCSEQAETGPCRAMISRWYFDVTEGKCAPFFYGGCGGNRNNFDTEEYCMAVCGSAMSQSLLKTTQEPLARDPVKLPTTAASTPDAVDKYLETPGDENEHAHFQKAKERLEAKHRERMSQVMREWEEAERQAKNLPKADKKAVIQHFQEKVESLEQEAANERQQLVETHMARVEAMLNDRRRLALENYITALQAVPPRPRHVFNMLKKYVRAEQKDRQHTLKHFEHVRMVDPKKAAQIRSQVMTHLRVIYERMNQSLSLLYNVPAVAEEIQDEVDELLQKEQNYSDDVLANMISEPRISYGNDALMPSLTETKTTVELLPVNGEFSLDDLQPWHSFGADSVPANTENEVEPVDARPAADRGLTTRPGSGLTNIKTEEISEVKMDAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIATVIVITLVMLKKKQYTSIHHGVVEVDAAVTPEERHLSKMQQNGYENPTYKFFEQMQN
Next, we will discuss Aβ, which is generally a cell surface receptor that carries out physiological tasks on the neuronal surface such as neurite outgrowth, neuronal adhesion, and axon genesis. Synaptogenesis is aided by interactions between amyloid precursor protein (APP) molecules on nearby cells [3]. Synaptogenesis is aided by interactions between APP molecules on nearby cells [3].
Through interactions between proteins, it also plays a role in transcriptional control and cell motility. Binding to APBB1-KAT5 can enhance transcriptional activation, whereas contact with Numb inhibits Notch signaling. coupled pathways that induce apoptosis, as those facilitated by G(o) and JIP. G(o)α ATPase activity is inhibited (by similarity).
It functions as a kinesin I membrane receptor, facilitating beta-secretase and presenilin 1 axonal trafficking (due to similarities).
As a kinesin I membrane receptor, which is involved in the anterograde transit of axons to synapses, it does this [4-5].
Engaging in the maintenance of copper balance and response to oxidative stress via the reduction of copper ions, the role of APP in copper homeostasis and neuronal health is multifaceted. In experimental settings, the interaction of copper with APP can trigger direct neuronal demise or be intensified through the oxidation of low-density lipoproteins (LDL) by Cu2+ ions. Additionally, APP influences the growth of neurites by interacting with extracellular matrix constituents such as heparin, and collagen types I and IV, which is facilitated by the presence of the BPTI domain in certain splice variants of the protein.
These BPTI-containing isoforms exhibit inhibitory effects on proteolytic activity. Furthermore, APP is known to initiate a signaling cascade that is AGER-dependent, leading to the phosphorylation and activation of p38 MAPK. This process subsequently promotes the internalization of amyloid-beta peptides, which can culminate in mitochondrial dysfunction within cortical neurons in culture. Lastly, APP serves as a provider of Cu2+ ions necessary for the function of GPC1, a protein crucial for the production and release of nitric oxide (NO). This copper ion provision is essential for the subsequent breakdown of heparan sulfate chains associated with GPC1, thereby maintaining the integrity and function of the cellular and extracellular environment.
The β-amyloid peptides exhibit a hydrophobic nature and function as metal-ion sequestrators, demonstrating the capability to reduce metals. They have a transient affinity for metals such as copper, zinc, and iron. In controlled in vitro conditions, these peptides are capable of reducing Cu2+ to Cu and Fe3+ to Fe2+. Among the variants, amyloid-beta protein 42 demonstrates a higher efficacy in the reduction process compared to its counterpart, amyloid-beta protein 40. These peptides interact with lipoproteins and apolipoproteins, namely E and J, within the cerebrospinal fluid (CSF) and with high-density lipoprotein (HDL) particles in plasma. This interaction plays a crucial role in mitigating the oxidation of lipoproteins, which is often catalyzed by metals. The presence of amyloid-beta protein 42 may also play a role in triggering the activation of mononuclear phagocytes within the brain, potentially leading to inflammatory reactions. Furthermore, β-amyloid peptides contribute to the aggregation of tau proteins and the phosphorylation mediated by TPK II. When in interaction with an overexpressed HADH2, these peptides can induce oxidative stress and neuronal toxicity. Additionally, they have been found to bind to GPC1, located within lipid rafts, which adds another layer to their multifaceted role in cellular processes.
In addition, numerous metal ion interaction sites are present, notably for copper and zinc ions. The sequestration of these metal ions, particularly copper, iron, and zinc, can lead to the formation of histidine bridges between amyloid-beta (Aβ) peptide molecules, culminating in the aggregation of Aβ-metal complexes. Copper demonstrates a significantly greater affinity for Aβ compared to other transient metals, and this attraction intensifies under conditions of low PH. The binding of extracellular zinc to amyloid precursor protein (APP) augments the interaction with heparin while concurrently suppressing the binding to collagen. This intricate interplay between metal ions and APP contributes to the modulation of various cellular processes, highlighting the importance of metal ion homeostasis in maintaining cellular function and integrity. Although there are 178 experimentally solved structures in the PDB structure database, these structures correspond to incomplete sequences.
Therefore, we will use AlphaFold2 to predict the structure corresponding β the complete sequence of amyloid. The tool we use is AlphaFold (AF for short), a revolutionary protein structure prediction software developed by DeepMind. Its main purpose is to predict how a protein folds in space and thus determine its three-dimensional structure. This is a critical step in understanding protein function and developing new drugs. The advent of AlphaFold2 is a great leap forward in the field of biology and computational biology, opening new avenues for the study of protein structures and drug development, as well as new challenges and research directions. It has the following components:
Input: The main input of the AlphaFold2 consists of the one-dimensional amino acid sequence of the protein. In addition, it can integrate various biological and experimental data to improve the accuracy of predictions.
Output: The output of AlphaFold2 is a three-dimensional structural prediction of the protein, including the relative position of its amino acids and the possible interactions between them.
2.2. The Model architecture of AlphaFold [6]
• Transformer model: AlphaFold uses a Transformer architecture like that used in natural language processing. This allows the model to efficiently capture amino acid relationships over long distances.
• Convolutional Neural Networks (CNNs): In some model components, AlphaFold also uses CNNs to process structural information, especially when dealing with distance plots and angular information of proteins
• Evolutionary information: MSA (Multiple Sequence Alignment): By aligning many related protein sequences, AlphaFold captures a wealth of evolutionary information that is critical to understanding how proteins fold.
• Deep learning architecture: AlphaFold derives the three-dimensional structure of proteins by combining features derived from MSA and extracted directly from the input sequence to predict the distance and interaction probability between amino acid residues.
• Evaluation and optimization (optimization functions) ---PLDDT (Predicted Local Distance Difference Test): AlphaFold provides researchers with valuable indicators of the quality of model predictions by using PLDDT scores to assess the confidence level of each amino acid in the structure predicted by the model.
• Loss function: AlphaFold uses a loss function that contains multiple different targets during training, for example, to optimize distance prediction, angle prediction, and other structural features to achieve comprehensive and accurate protein structure prediction.
By integrating the above components, AlphaFold not only accurately predicts protein structure, but also provides a powerful tool for biological research by understanding how proteins fold and how they interact with each other.
2.3. AlphaFold2’s Advantages [7]
• High accuracy: It is far more accurate than previous methods in predicting protein structure. The CASP14 evaluation provides a detailed comparison of leading structural prediction methods. AlphaFold was the highest-ranked methodology, with a median GDT (Global Distance Test) score of 92.4 for all targets and a median GDT score of 87.0 for the challenging free modelling category, while the second-best methods in these categories were 72.8 and 61.0, respectively. Structural biologists more commonly express the similarity between two protein structures by first superimposing the structures in an optimal manner and then calculating the root-mean-square distance (RMSD) between the Cα atoms of the equivalent residues. Taking the median RMSD-Cα at the best predicted 95% residue reduces the effects of flexible tails and crystal packing artifacts. On this metric, the median distance between AlphaFold’s CASP14 prediction and the experimental model is 0.96 Å, compared to 2.83 Å for the suboptimal method.
• Wide applicability: Ability to process multiple types of protein sequences.
• Open source: DeepMind made AlphaFold’s code and model publicly available after its research paper was published, allowing the scientific community to use and improve the tool.
However, it also has the following limitations:
• Despite the great success of AlphaFold2 in protein structure prediction, its predictive performance may still be limited for some particularly complex proteins or protein complexes.
• Static structure: AlphaFold2 predicts the static structure of proteins, but proteins change dynamically within the cell.
• AlphaFold has not been validated to predict the impact of mutations. Given the sequence containing unstable point mutations, AlphaFold is not expected to produce unfolded protein structures.
• AlphaFold’s predictive capabilities are primarily focused on the structure of proteins as they are typically represented in the Protein Data Bank (PDB). While it does not forecast the positions of non-protein elements such as cofactors, metals, ligands, ions, nucleic acids, or post-translational modifications that are part of experimental setups, AlphaFold is adept at modeling the protein structure. Consequently, when proteins are associated with ions (for instance, sites that bind zinc) or cofactors (like those with side chain configurations that align with heme attachment), the software often accurately predicts the protein’s backbone and side chain conformation in line with the anticipated structure.
• It is essential to note that AlphaFold’s predictive models do not account for the influence of ligands, ions, covalent alterations, or environmental factors. As a result, the system may not always capture the full spectrum of structural nuances that are contingent upon these elements. This limitation underscores the need for complementary experimental data to fully comprehend the intricate details of protein structures that are influenced by such factors [8].
3. Results
3.1. AlphaFold predicted structures of Aβ [9]
Here’s how we predicted the complete structure of Aβ sequence with AlphaFold from four perspectives:
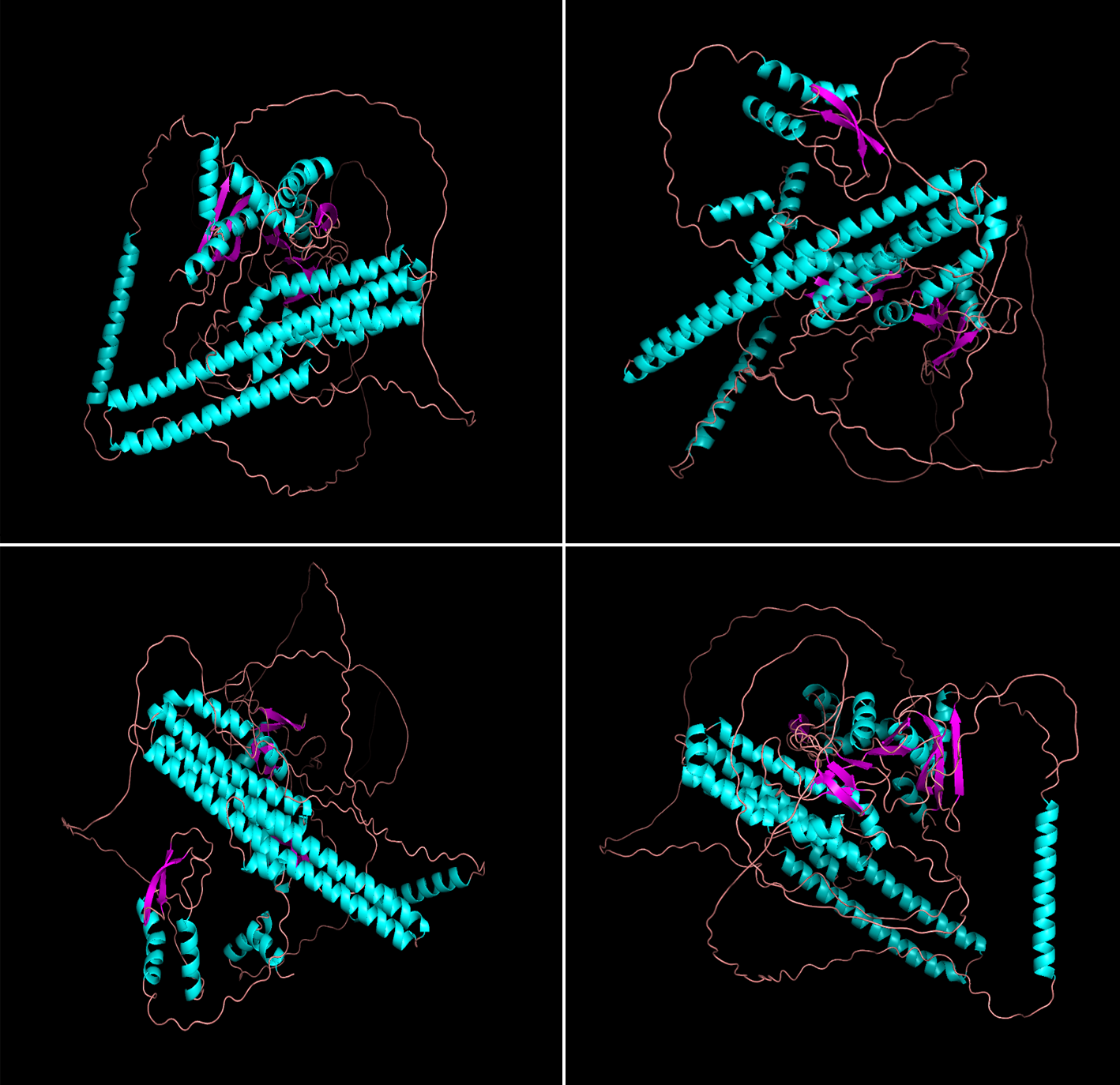
Figure 1. The four predicted structures of βamyloid generated by PyMOL.
In this model, the compact core parts are mainly blue and dark purple, and the confidence read is high, while the sequence color connecting the parts connecting each domain is light red, and the confidence is low. The overall confidence level is a step up from the previous model.
The following is a brief description of the sequence of this protein:
The middle part of the 700-726 sequence has high confidence, which is the α helix part, and the connecting part at both ends has low confidence, which is the β chain. This peptide chain is susceptible to γ-secretase cleavage, and its main role is to interact with PSEN1.
The 425-489 sequence is partially a α helix with high confidence, but there is no binding site in the current study.
Sequences 744-766 are partially of low confidence, with part of 744-752 being α helix strucon with KIF5B and anterograde transport of axons.tures associated with G(o)-α interactions, and parts 752-766 being β chain structures for interacti.
The 281-290 sequence is partially of low confidence, and the 288-292 parts is a α helix with no binding site, and the function is not well understood.
The 388-419 sequence is partially confident, is a α helix, and is mainly used for binding to heparin.
The 427-480 sequence is partially confident, with α helix, no binding site, and its function is unclear.
The 521-549 sequence is partially confident, is a α helix, and binds to collagen.
The 521-549 sequence moiety is highly confident, and it is a α helix moiety, which belongs to the GFLD subdomain of the protein.
Building on the foundation laid in the previous section, where the β-amyloid monomer’s target structure was approximated using AlphaFold, we proceeded to discern regions that are pivotal for structure-based drug design. This was followed by the application of the Protein-Ligand Interaction Profiler (PLIP) to model the non-covalent interactions upon ligand binding, thereby elucidating key binding sites that correspond to the crucial crystal structure.
As we progress further, we shift our focus to the structural analysis of the 5NX1 complex. Within this context, we continue to harness PLIP’s capabilities to delve into the non-covalent interactions between the amyloid and K6 proteins within the β-amyloid system, aiming to enhance our understanding of the molecular underpinnings that govern these interactions.
3.2. The structural analysis of the crystal structure of Aβ binding with its ligand
Structure of 5NX1 complex and non-covalent bonding conditions predicted by PLIP:
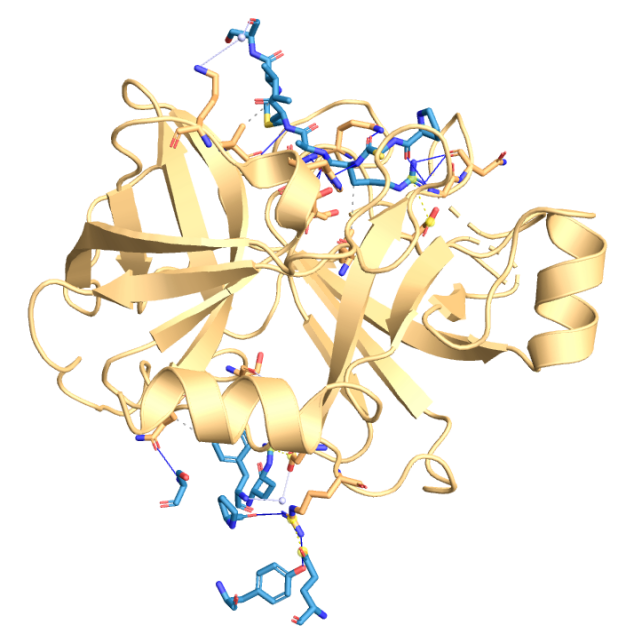
Figure 2. (from PLIP) Using PLIP to predict binding sites in 5NX1 with interacting chains: B, C, D [10].
Non-covalent bonding was not predicted in Aβ monomers, so we used the PDB complex of 5NX1 to detect some non-covalent bonds:
Table 1. Hydrophobic Interactions in 5NX1.
Index | Residue | AA |
1 | 15C | ARG |
2 | 18C | ILE |
3 | 34D | PHE |
Table 2. Water Bridges in 5NX1.
Index | Residue | AA |
1 | 15B | ARG |
2 | 19C | ILE |
3 | 34D | PHE |
Table 3. Hydrogen Bonds in 5NX1.
Index | Residue | AA |
1 | 13C | PRO |
2 | 14C | CYS |
3 | 15C | ARG |
4 | 15C | ARG |
5 | 15C | ARG |
6 | 15C | ARG |
7 | 15C | ARG |
8 | 15C | ARG |
9 | 16C | ALA |
10 | 17C | MET |
11 | 19D | SER |
12 | 22D | TYR |
13 | 22D | TYR |
14 | 32D | PRO |
Table 4. Salt Bridges in 5NX1.
Index | Residue | AA |
1 | 15B | ARG |
2 | 15C | ARG |
3 | 27D | GLU |
Table 5. Summary of non-covalent bonds in 5NX1.
Type of non-covalent bones | Quantity | Positions |
Hydrophobic Interactions | 3 | 15C, 18C, 34D |
Water Bridges | 3 | 15B, 19C, 34D |
Hydrogen Bonds | 14 | 13C, 14C, 15C, 16C, 17C, 19D, 22D, 32D |
Salt Bridges | 3 | 15B, 15C, 27D |
4. Conclusion
The present study has reaffirmed the significant role of amyloid-beta protein (Aβ) in the etiopathogenesis of Alzheimer’s disease (AD), with a wealth of data underscoring its central involvement. The association of Aβ with AD pathogenesis has directed our focus towards this protein as a prime therapeutic target. Leveraging the computational power of AlphaFold, we have achieved a high degree of accuracy in predicting the structural arrangement of Aβ, including the disposition of beta-sheets and alpha-helices, and their interactive dynamics, which are pivotal in discerning potential toxic conformations. Such insights are essential for elucidating the pathological mechanisms underpinning AD and for the formulation of targeted therapeutic interventions.
Building upon the enhanced three-dimensional structural models of Aβ provided by AlphaFold, alongside predictions of drug molecule interactions with the receptor (RSBDD), we have delineated a pathway for the design of drugs with heightened affinity and selectivity.
However, it is important to acknowledge that the predictive accuracy of AlphaFold, despite its laudable performance in structural prognostication, necessitates empirical validation. The intrinsic structure of Aβ may be subject to modulation by in vivo post-translational modifications, such as glycosylation and phosphorylation, and by interactions with a myriad of other proteins, all of which may potentially induce structural and functional alterations.
Through Protein-Ligand Interaction Profile (PILP) analysis of the 5NX1 complex, an in-depth understanding of the interactions between beta-amyloid protein and K6 protein was achieved, revealing the precise locations and modes of their binding, including hydrogen bonds, hydrophobic interactions, and salt bridges. This contributes to the comprehension of the structural characteristics of the protein’s active site and ligand-binding site, offering insights for the discovery of novel therapeutic targets and drugs. Researchers can design new molecules with higher affinity and selectivity, facilitating the development and optimization of new drugs targeted at Alzheimer’s disease.
Therefore, the Aβ structure and bonding predictions provided by AlphaFold and PLIP should be used as the initial reference point, and further experimental studies are essential to confirm and improve whether the initial reference point can be used in the treatment of Alzheimer’s disease.
References
[1]. Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., & Ashe, K. H. (2005). Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nature neuroscience, 8(1), 79-84.
[2]. UniProt Consortium. (2019). UniProt: a worldwide hub of protein knowledge. Nucleic acids research, 47(D1), D506-D515.
[3]. Baumkötter, F., Schmidt, N., Vargas, C., Schilling, S., Weber, R., Wagner, K., ... & Kins, S. (2014). Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. Journal of Neuroscience, 34(33), 11159-11172.
[4]. Satpute-Krishnan, P., DeGiorgis, J. A., Conley, M. P., Jang, M., & Bearer, E. L. (2006). A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proceedings of the National Academy of Sciences, 103(44), 16532-16537.
[5]. Seamster, P. E., Loewenberg, M., Pascal, J., Chauviere, A., Gonzales, A., Cristini, V., & Bearer, E. L. (2012). Quantitative measurements and modeling of cargo–motor interactions during fast transport in the living axon. Physical biology, 9(5), 055005.
[6]. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., ... & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583-589.
[7]. Marx, V. (2022). Method of the Year: protein structure prediction. Nature methods, 19(1), 5-10.
[8]. Terwilliger, T. C., Liebschner, D., Croll, T. I., Williams, C. J., McCoy, A. J., Poon, B. K., ... & Adams, P. D. (2024). AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nature Methods, 21(1), 110-116.
[9]. Krämer, K. (2024). AI & robotics briefing: AlphaFold predicts thousands of possible psychedelics. Nature.
[10]. Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F., & Schroeder, M. (2015). PLIP: fully automated protein–ligand interaction profiler. Nucleic acids research, 43(W1), W443-W447.
Cite this article
Fang,T. (2024). Utilizing deep learning for alzheimer's disease treatment: Targeting β-amyloid for therapeutic intervention. Theoretical and Natural Science,49,66-73.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., & Ashe, K. H. (2005). Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nature neuroscience, 8(1), 79-84.
[2]. UniProt Consortium. (2019). UniProt: a worldwide hub of protein knowledge. Nucleic acids research, 47(D1), D506-D515.
[3]. Baumkötter, F., Schmidt, N., Vargas, C., Schilling, S., Weber, R., Wagner, K., ... & Kins, S. (2014). Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. Journal of Neuroscience, 34(33), 11159-11172.
[4]. Satpute-Krishnan, P., DeGiorgis, J. A., Conley, M. P., Jang, M., & Bearer, E. L. (2006). A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proceedings of the National Academy of Sciences, 103(44), 16532-16537.
[5]. Seamster, P. E., Loewenberg, M., Pascal, J., Chauviere, A., Gonzales, A., Cristini, V., & Bearer, E. L. (2012). Quantitative measurements and modeling of cargo–motor interactions during fast transport in the living axon. Physical biology, 9(5), 055005.
[6]. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., ... & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583-589.
[7]. Marx, V. (2022). Method of the Year: protein structure prediction. Nature methods, 19(1), 5-10.
[8]. Terwilliger, T. C., Liebschner, D., Croll, T. I., Williams, C. J., McCoy, A. J., Poon, B. K., ... & Adams, P. D. (2024). AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nature Methods, 21(1), 110-116.
[9]. Krämer, K. (2024). AI & robotics briefing: AlphaFold predicts thousands of possible psychedelics. Nature.
[10]. Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F., & Schroeder, M. (2015). PLIP: fully automated protein–ligand interaction profiler. Nucleic acids research, 43(W1), W443-W447.