







1. Introduction
Poly ADP-Bibose polymerase (PARP) is a ribozyme found in eukaryotic cells and plays an important role in involving DNA repair and maintaining genomic stability [1]. PARP-1 is the most important member involved in different DNA repair pathways, including single-strand DNA break repair (SSBR), nucleotide excision repair (NER), Alternative non-homologous end junctions (alt-NHEJ), and homologous recombination (HR).
Studies have shown the overexpression of PARP in tumors, inflammation-related diseases, autoimmune diseases, neurodegenerative diseases, and metabolic stress [2]. So far, the role of PARP in tumors has been best studied, and the inhibition of PARP activity in tumor cells can reduce DNA repair in tumor cells, thus promoting tumor cell death. Therefore, PARP has become an important target for tumor drug development [2]. In tumor cells with homologous recombination repair (HRR) defects, the resulting DNA double-stranded breaks are effective in mass killing of cancer cells if PARP activity is inhibited. Stated differently, PARP inhibitors can work in concert to have deadly effects on tumors that carry mutations in the gene that makes people susceptible to breast cancer (BRCA). BRCA mutations were first identified in ovarian and breast cancers, and prostate and pancreatic cancers were also reported to have some relationship with BRCA gene mutations [2]. Therefore, PARP inhibitors are expected to be ideal drugs for cancer therapy with wide research implications [2].
DNA damage responses in BRCA1/2 mutated breast and ovarian cancers can be specifically targeted by PARP inhibitors, the first approved cancer drugs that specifically aim for these reactions [3]. In cancer patients with BRCA1/2 mutations or genomic instability, PARP inhibitors are also permitted in the use of bevacizumab as a combination [4]. Currently, various PARP inhibitor drugs are available on the market, including Olaparib, Niraparib, Talazoparib and Rucaparib, and their main mechanism of action is to inhibit the activity of PARP. Despite the different action targets of these inhibitors, most drugs will target both PARP-1 and PARP-2, thus having some limitations in terms of selectivity. Note that, although PARP-2 expression is relatively low, its absence may lead to a range of adverse effects. Studies have shown that by binding the noncatalytic site of PARP, it is expected to improve the selectivity of PARP inhibitors to improve the efficacy and prolong the progression-free survival of patients, thus providing more effective and safer drug options for cancer treatment. Besides, drug resistance is unavoidable, and resistance is a possibility for approximately 40-70% of patients [5].
2. The structure of PARP
Six domains make up the modular structure of PARP-1 (Figure 1): the two zinc-binding domains, Zn 1 and Zn 2, allow PARP-1 to detect particular DNA configurations, while the Zn 1 and Zn 3 domains promote PAR synthesis of WGR-CAT fragments when DNA is present [6]. Automodification domain (AD) has major sites for self-modification [6]. The central auto-modified region contains a breast cancer susceptibility gene carboxyl-terminal (BRCT) domain of the breast cancer susceptibility gene and flanking fragments containing glutamate or lysine residues that can serve as automatic ADP ribosylation sites [6].
The tryptophan-glycine-arginine domain (WGR) is the center of the PARP-binding complex with DNA [6]. The PARP-1 catalytic domain (CAT) contains the ART subdomain (ADP-ribosyl transferases, ARTs) consisting of a bound nicotinamide adenine dinucleotide (NAD+) is composed of the donor site of the β-α-loop-β-α characteristic motif, the acceptor site of the ADP-ribose chain extension, and the helical domain (HD) [7]. The HD consists of six α-helices, forming a hydrophobic core that contributes to the folded of the ART domain [7].
Zn 1, Zn 3, and WGR-CAT are the structural domains required for the functional activity of PARP [6].
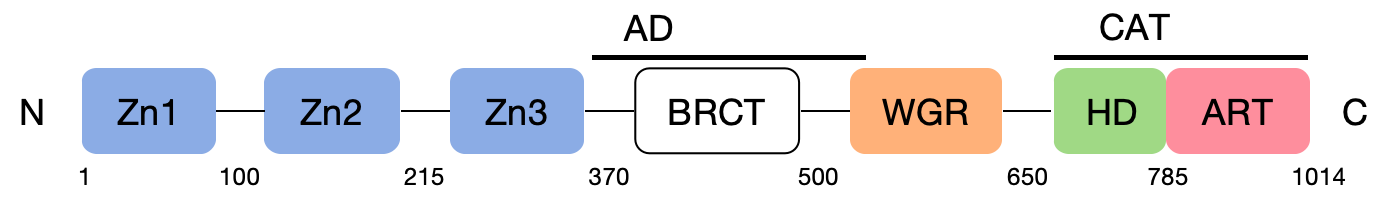
Figure 1. The structure of PARP.
3. The repair mechanism of PARP
After DNA damage occurs, the Zn region of PARP-1 rapidly recognizes and binds the damage site [2]. The binding of PARP to a DNA incision can cause a temporary arrest of cellular activity, thereby allowing the proximity of DNA repair enzymes to the site of damage [8]. At this point, PARP changes its conformation and thus activates CAT [2]. CAT catalyzes NAD+, which decomposes into nicotinamide and ADP ribosidine, and then uses the resulting ADP ribose as the substrate to phosphorylate the receptor proteins involved in repair to form polyadenosine diphosphate ribose (poly (ADP-ribose), PAR) [2]. This process, called PAR location (Figure 2), mediates the recruitment of single-stranded DNA repair effectors [9]. Histones, PARP-1, and a few other DNA repair proteins are examples of the receptor proteins. Because of their high negative charge, the proteins altered by PAR polymer have the ability to attract some key players in the BER-SSB cascade, including XRCC 1. Non-covalently interacting with PAR, other proteins involved in chromosomal spatial structure development, DNA repair, and replication can also contribute to DNA damage repair [10].
The dissociated PAR is cleaved by polyadenosine diphosphate ribose hydrolase, and the cleaved ADP ribose can be reused for NAD+ synthesis, Whereas the monomer-reformed PARP-1 can be reactivated [2].
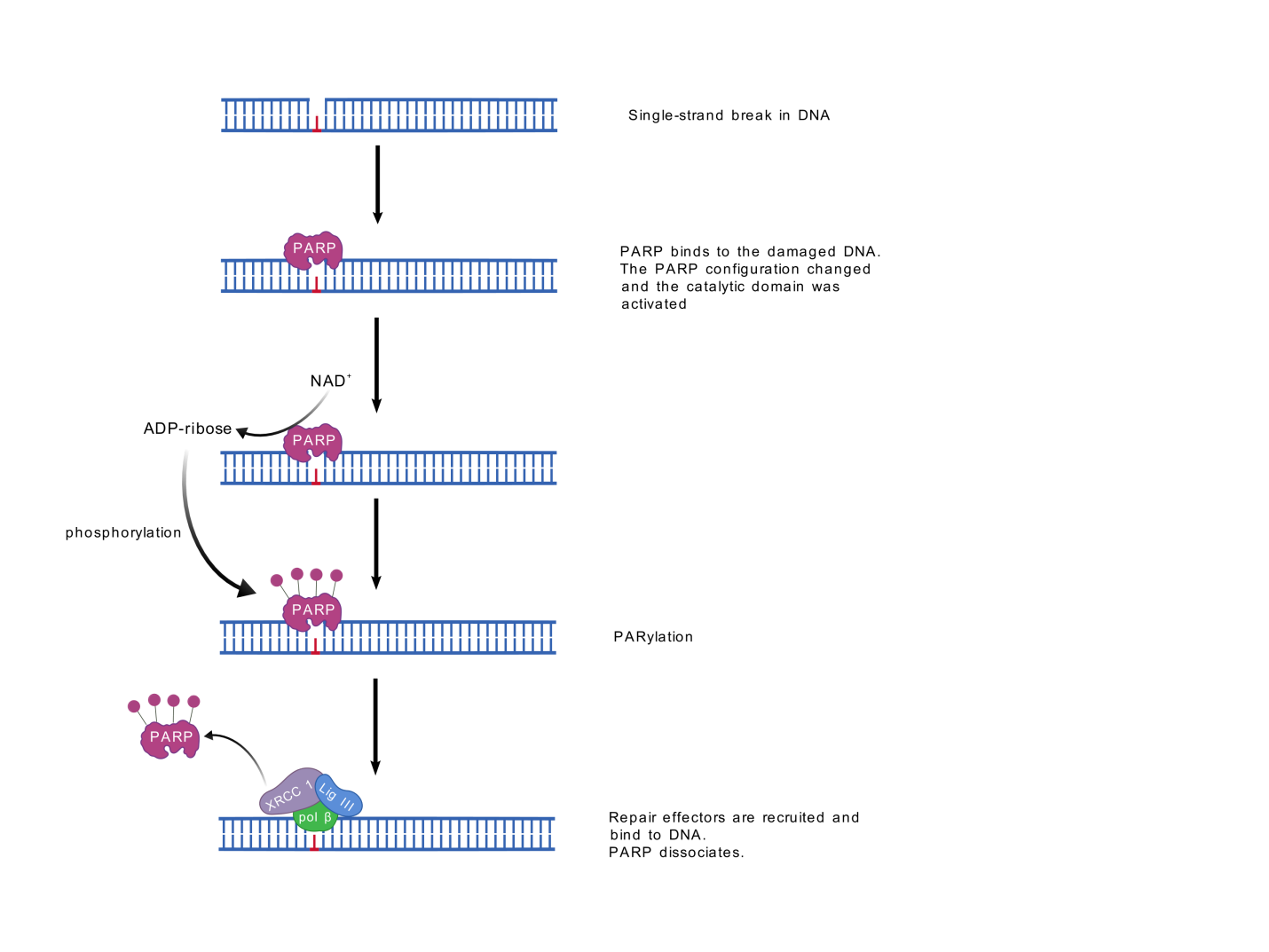
Figure 2. PARP recruitment of proteins.
4. Mechanism of the PARP inhibitors
The term synthetic lethality describes a situation where two non-lethal gene mutations occur independently without affecting cell viability, but when they occur together, they produce cell death [11]. Decades later, it was found that cells carrying BRCA mutations were 1,000 times more sensitive to PARP-1 inhibition than wild-type cells [12], providing a theoretical basis for defining PARP-1 as a synthetic lethal partner of BRCA. This makes it possible to test PARP inhibitors and other drugs that target DNA repair pathways in BRCA 1 or 2 mutated cancers [13].
When DNA single-strand breaks (SSB) occur in cells, PARP begins to functionally repair DNA. However, in the presence of PARP inhibitors, PARP inhibitors bind to PARP, inhibit PAR lation, and trap inactivated PARP on the DNA, thus blocking the replication fork, leading to replication fork collapse and creating double-strand breaks [9] (Figure 3). The PARP-DNA complex that was captured is more cytotoxic than SSBs that were not reattached after PARP inactivation, which indicates that PARP inhibitors are toxic agents that trap PARP enzymes on DNA in part [14].
In cells, there are two ways to repair DNA double-strand breaks (DSB): homologous recombination repair and non-homologous recombination repair. In cells with normal repair by homologous recombination, DNA is able to be repaired normally and cells can survive. However, the conventional belief is that tumor cells experience increased genomic instability due to loss of HR repair, which causes more mutations to be produced and cancer to progress [15]. In cells defective in homologous recombination repair are cells with BRCA mutations, DSB cannot be efficiently repaired, so severe damage to DNA will cause cell death. DSB can be repaired by a nonhomologous end-joining pathway. However, this misvulnerability of the template-independent repair pathway ultimately leads to tumor cell death [9] (Figure 3). That is to say, cell death occurs due to massive chromatid aberrations when the error-prone alt-NHEJ repair mechanism is unable to repair chromatid breaks caused by simultaneous inhibition of PARP-1 function in BRCA1 or 2 defects [16].
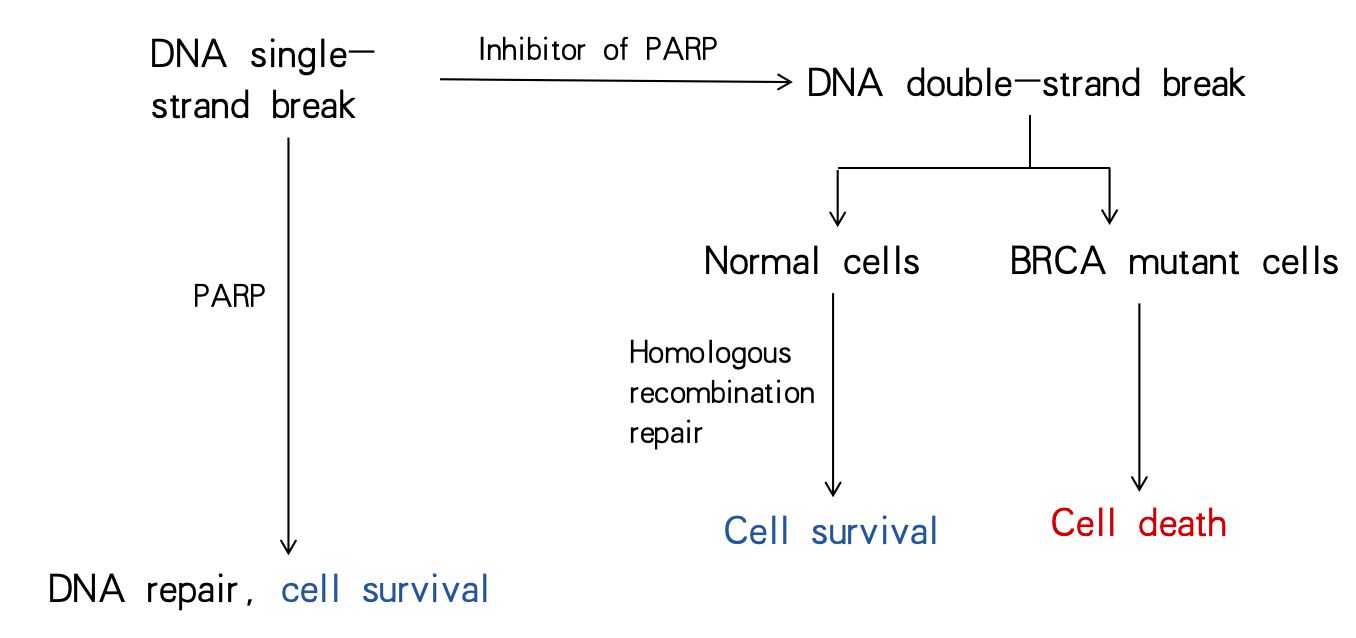
Figure 3. Cell fate before and after PARP inhibition.
5. Design of the PARP Inhibitor drugs
The primary intracellular generator of PAR, PARP-1, is activated by attaching to the DNA damage site [17]. The catalytic activation of PARP-1 is binding to DNA through the N-terminal zinc finger (ZnF), HD folding, and NAD+, which is a multistep process of binding to the catalytic pocket and PAR catalysis [17].
The current mechanism of action of the mainstream and effective PARP inhibitors is mainly through CAT and NAD+ in the catalytic domain of PARP to form a competitive relationship between them. Given the high conservation of the catalytic pocket of the PARP family, drugs relying solely on this design are deficient in terms of specificity. Although the initially developed drugs can be well combined with PARP, their key properties such as drug potency and water solubility still need to be further improved. In order to ensure the safety and effectiveness of the drug, PARP inhibitors should introduce other functional groups besides focusing on competing sites.
5.1. The binding Site for the PARP Inhibitors
The first PARP-1 inhibitor was nicotinamide itself, followed by 3-aminobenzamide (3-AB) [17] (Figure 4). The cytotoxic effects of the DNA methylator dimethyl sulfate were enhanced by 3-AB in the 1980s [18]. All subsequent developed PARP-1 inhibitors contained a nicotinamide or benzamide pharmacophore and were associated with NAD+ competitive catalytic pocket of PARP [17]. The kinetic site is bound to by PARP-1 inhibitors through the formation of hydrogen bonds with Gly, Ser, and Glu, as well as by the formation of hydrophobic packing interactions with two Tyr residues in the nicotinamide-binding pocket [17].
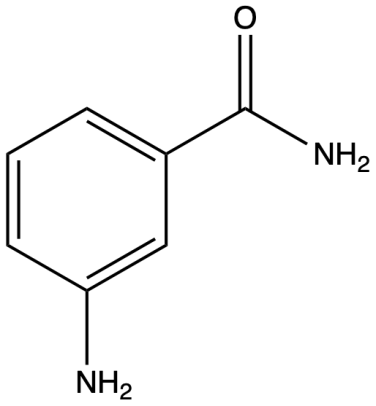
Figure 4. Structure of 3-aminobenzamide (3-AB).
Three hydrogen bonds are established in the nicotinamide subsite of the pocket in NAD+, including two from Gly 863 and one from Ser 904 [19] (Figure 5). The posterior wall of the nicotinamide subsite bounded by Ala 898 and Lys 903 forms a tight small pocket that is just large enough to accommodate small substituents (e. g. CH3, F, Cl) on the benzamide-containing ring [19] (Figure 5).
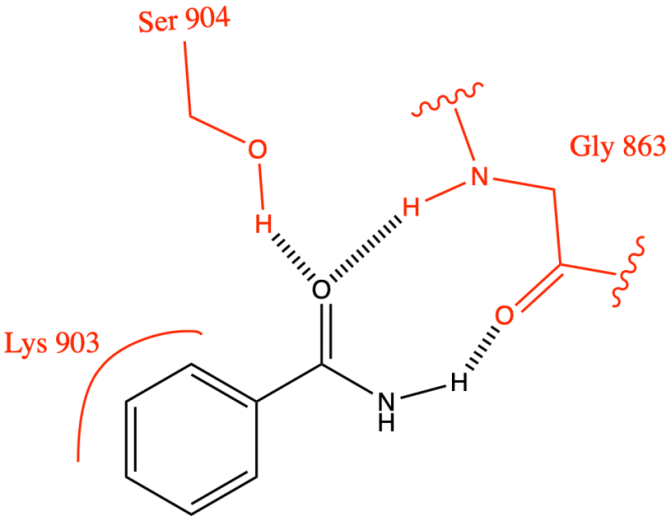
Figure 5. NAD+ catalytic pocket for amino acid interactions.
5.2. Design and function of the functional groups of different inhibitors
5.2.1. Olaparib
The chemical name of Olaparib is 1- (cyclopromonyl) -4- [5- [(3,4-dihydrogen-4-oxygen-1-phthalazine) methyl] -2-fluorbenzoyl] piperazine [20], chemical structures as shown in Figure 6a.
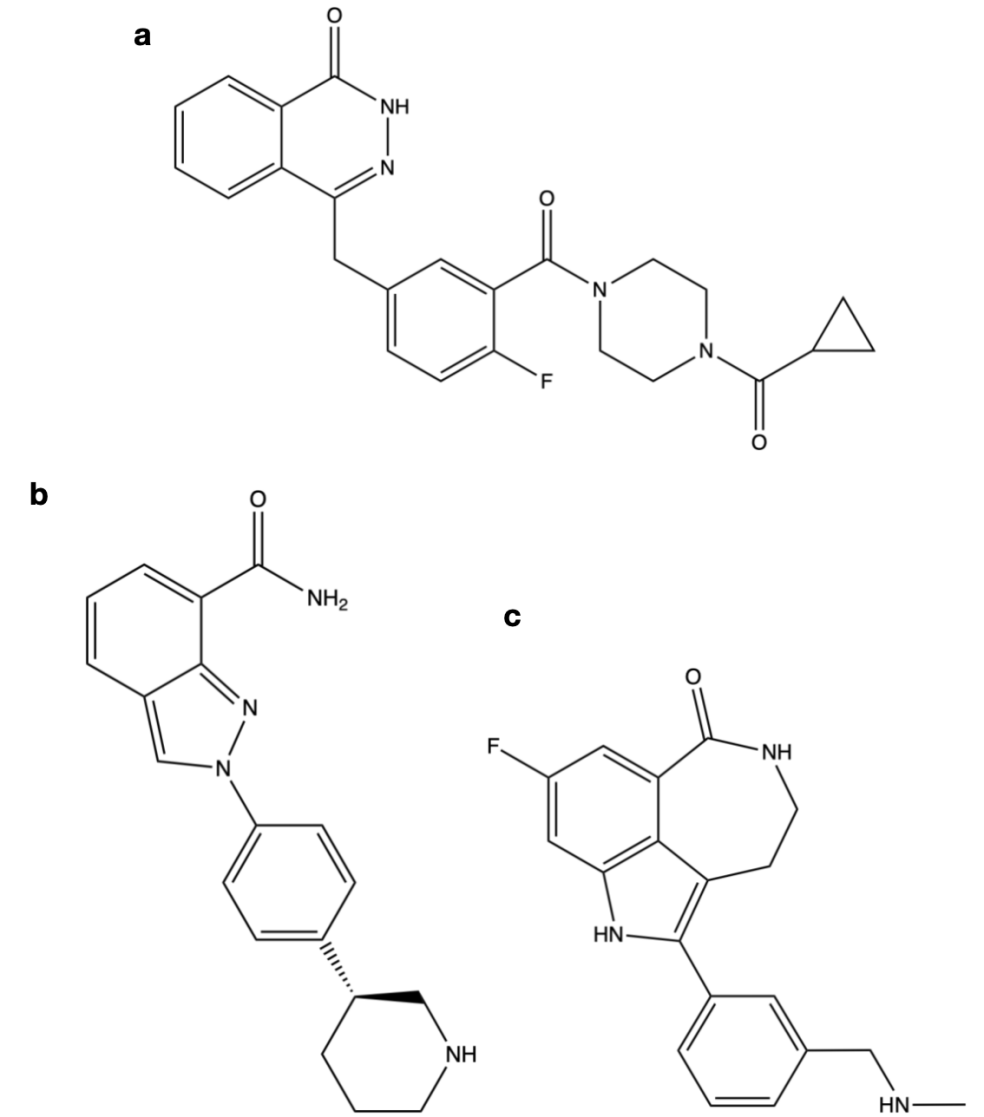
Figure 6. Chemical structure diagram of the PARP inhibitor.
By restricting the degree of freedom of the amide moiety, Olaparib secures the aryl amide in another loop and promotes the inhibitory potency of PARP-1 [19]. The benzyl group enhances cell activity, while the diacylpiperazine segment retains enzyme potency (IC 50 = 5 nM) and raises cell death (PF 50 = 25.8), all while boosting solubility to 81 [19]. Cyclopropyl group can effectively improve the oral bioavailability [19].
5.2.2. Niraparib
The chemical name of Niraparib is 2- [4- [(3S) -piperidine-3-group] phenyl] azole-7-carboxamide, and the chemical structure is shown in Figure 6b.
Niraparib initially became a PARP inhibitor candidate for its superior pharmacokinetic properties and enzyme and cellular potency [19]. Its aryl group as a solubilizing group leads to a general improvement in enzymatic and cellular potency [19]. (S) -piperidine showed better cellular potency and BRCA selectivity than (R) -piperidine, while amine 2 improved solubility and pharmacokinetic parameters [19].
5.2.3. Rucaparib
The chemical name of Rucaparib is 8-fluorine-2- {4- [(methine) methyl] phenyl} -1,3,4,5-tetrahydrogen-6H-nitrogen and [5,4,3-CD] index-6-ketone ((1S, 4R) -7,7-dimethyl-2-oxygen duplex [2.2.1] ethylene-1-group) mesylate, and the chemical structure is shown in Figure 6c.
When amide NH is bound to imidazole nitrogen, it becomes an intramolecular hydrogen bond acceptor. Locking the procamides into the geometry that is the most conducive to PARP-1 binding, similar to bicyclic and tricyclic lacamides, is what causes the PARP-1 binding to be most effective [19].
The aryl group interacts with the π - π of Tyr 889 and Tyr 907 in PARP-1 at its nicotinamide-binding site [19]. The free carboxamide is restricted within the seven-membered ring, producing the same result as the intramolecular hydrogen bond, forming the tricyclic imidazololactam [19]. The secondary amine improved the water solubility, inhibitory potency, and cell permeability of the arylamide core [19]. Indo NH forms a water-mediated hydrogen bond with Glu 988, while the dimethylamine group interacts with Asp 766 [19].
5.3. DNA capture
PARP-1 capture is thought to be dependent on allosteric changes in the PARP-1-DNA binding domain, and the allosteric changes described by a PARP inhibitor with NAD+ induction of D loop binding at the outer boundary of the site [17]. PARP capture can take place in areas where DNA strands are broken, where topoisoselectase I (TOP 1) has been used, and where okazaki fragment intermediates of DNA replication are unrelated [17]. The inhibition of its catalytic activity renders PARP-1 unable to dissociate from DNA upon being trapped [17].
DNA capture efficiency: Talazoparib>> Niraparib> Olaparib = Rucaparib>> Veliparib.
5.4. PARP inhibitor specificity
Veliparib topped the list of clinically relevant inhibitors for its selectivity of PARP-1 / 2 inhibitors, followed by Niraparib [17]. The unique water-mediated hydrogen bond formed between PARP-1/2 and residues of the regulatory subdomain (D766 in PARP-1) is what determines their selectivity, and it is conserved in PARP-1/2, not in other PARPs [17]. Veliparib has a selectivity for PARP-1/ 2 that exceeds 100 times the other family members', while Olaparib and Talazoparib only have a selectivity of 15 to 20 times higher [17]. Rucaparib is the PARP-1 inhibitor that is least selective in pharmacology, blocking PARP-1 while also stopping the single (ADP-ribosyl) transferases PARP-3, PARP-4, PARP-10, PARP-15, and PARP-16 [17].
6. Conclusion
The screening and marketing of PARP inhibitor drugs is breakthrough for the treatment of BRCA-mutant cancers, and their unique chemical structures confer different key parameters such as DNA capture efficiency, enzyme and cellular potency, and permeability. Therefore, according to the specific situation of the patient, medical institutions can develop personalized treatment plans to maximize the treatment effect. Moreover, based on the mechanism of action of PARP inhibitors, these drugs are expected to produce synergistic effects with other drugs, providing new possibilities for cancer therapy.
After an exhaustive analysis of drug design concepts and chemical architecture, the drug molecule usually contains a nicotinamide or benzamide as a core structure bound to the catalytic domain conserved in the PARP family. Beyond this, other parts of the drug molecule are mostly devoted to optimizing drug properties. However, so far, the designed drugs have yet to achieve the ideal PARP-1 selectivity, where PARP-2 is often identified simultaneously as a non-specific target. Recent studies have shown that enhanced targeting to PARP noncatalytic sites may help to enhance drug selectivity. Therefore, in the future research work, the precise design and screening of highly selective drugs will become the core strategy to weaken and even solve the current PARP targeted adverse drug reactions.
References
[1]. Nie Lu 2019 Oral PARP inhibitors—Talazoparib Journal of Clinical Pharmacotherapy 17 02 67-70
[2]. Dan W Xiaoli H Huajian Z Jia'an S Wei H Jiankang Z 2022 Progress of selective PARP-1 inhibitors Modern Applied Pharmacy in China 39 20 2697-2706
[3]. Rose M Burgess J T O'Byrne K Richard D J & Bolderson E 2020 PARP Inhibitors: Clinical Relevance Mechanisms of Action and Tumor Resistance Frontiers in cell and developmental biology 8 564-601
[4]. Paik J 2021 Olaparib: A Review as First-Line Maintenance Therapy in Advanced Ovarian Cancer Targeted oncology 16(6) 847–856
[5]. Kim D & Nam H J 2022 PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them International journal of molecular sciences 23(15) 8412
[6]. Langelier M F Planck J L Roy S & Pascal J M 2012 Structural basis for DNA damage-dependent polyADP-ribosylation by human PARP-1 Science 3366082 728-732
[7]. Steffen J D Brody J R Armen R S & Pascal J M 2013 Structural Implications for Selective Targeting of PARPs Front Oncol 3 301
[8]. White A W Almassy R Calvert A H Curtin N J Griffin R J Hostomsky Z Maegley K Newell D R Srinivasan S & Golding B T 2000 Resistance-modifying agents 9 Synthesis and biological properties of benzimidazole inhibitors of the DNA repair enzyme polyADP-ribose polymerase J Med Chem 4322 4084-4097
[9]. Cortesi L Rugo H S & Jackisch C 2021 An Overview of PARP Inhibitors for the Treatment of Breast Cancer Target Oncol 163 255-282
[10]. Zhen Q Yiyuan W Lusong L Lai W Min W 2014 Anti-tumor targeted drugs for PARP Chinese Journal of New Drugs 23 17 2052-2056
[11]. DOBZHANSKY T 1946 Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura Genetics 31(3) 269–290
[12]. Farmer H McCabe N Lord C J Tutt A N Johnson D A Richardson T B Santarosa M Dillon K J Hickson I Knights C Martin N M Jackson S P Smith G C & Ashworth A 2005 Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy Nature 434(7035) 917–921
[13]. Ferrara R Simionato F Ciccarese C Grego E Cingarlini S Iacovelli R Bria E Tortora G & Melisi D 2018 The development of PARP as a successful target for cancer therapy Expert review of anticancer therapy 18(2) 161–175
[14]. Murai J Huang S Y Das B B Renaud A Zhang Y Doroshow J H Ji J Takeda S & Pommier Y 2012 Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors Cancer research 72(21) 5588–5599
[15]. D'Andrea A D 2018 Mechanisms of PARP inhibitor sensitivity and resistance DNA repair 71 172–176
[16]. Patel A G Sarkaria J N & Kaufmann S H 2011 Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells Proceedings of the National Academy of Sciences of the United States of America 108(8) 3406–3411
[17]. Slade D 2020 PARP and PARG inhibitors in cancer treatment Genes Dev 345-6 360-394 Ferraris D V 2010 Evolution of polyADP-ribose polymerase-1 PARP-1 inhibitors From concept to clinic J Med Chem 5312 4561-4584
[18]. Purnell M R & Whish W J 1980 Novel inhibitors of poly(ADP-ribose) synthetase The Biochemical journal 185(3) 775–777
[19]. Ferraris D V 2010 Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors From concept to clinic J Med Chem 53(12) 4561-4584
[20]. Benchuan C 2015 olaparib a new drug for the treatment of advanced ovarian cancer Medical Review 34 06 846-849
Cite this article
Zhang,Y. (2024). PARP inhibitor drugs and their targeted therapy for BRCA gene mutations. Theoretical and Natural Science,49,98-105.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Nie Lu 2019 Oral PARP inhibitors—Talazoparib Journal of Clinical Pharmacotherapy 17 02 67-70
[2]. Dan W Xiaoli H Huajian Z Jia'an S Wei H Jiankang Z 2022 Progress of selective PARP-1 inhibitors Modern Applied Pharmacy in China 39 20 2697-2706
[3]. Rose M Burgess J T O'Byrne K Richard D J & Bolderson E 2020 PARP Inhibitors: Clinical Relevance Mechanisms of Action and Tumor Resistance Frontiers in cell and developmental biology 8 564-601
[4]. Paik J 2021 Olaparib: A Review as First-Line Maintenance Therapy in Advanced Ovarian Cancer Targeted oncology 16(6) 847–856
[5]. Kim D & Nam H J 2022 PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them International journal of molecular sciences 23(15) 8412
[6]. Langelier M F Planck J L Roy S & Pascal J M 2012 Structural basis for DNA damage-dependent polyADP-ribosylation by human PARP-1 Science 3366082 728-732
[7]. Steffen J D Brody J R Armen R S & Pascal J M 2013 Structural Implications for Selective Targeting of PARPs Front Oncol 3 301
[8]. White A W Almassy R Calvert A H Curtin N J Griffin R J Hostomsky Z Maegley K Newell D R Srinivasan S & Golding B T 2000 Resistance-modifying agents 9 Synthesis and biological properties of benzimidazole inhibitors of the DNA repair enzyme polyADP-ribose polymerase J Med Chem 4322 4084-4097
[9]. Cortesi L Rugo H S & Jackisch C 2021 An Overview of PARP Inhibitors for the Treatment of Breast Cancer Target Oncol 163 255-282
[10]. Zhen Q Yiyuan W Lusong L Lai W Min W 2014 Anti-tumor targeted drugs for PARP Chinese Journal of New Drugs 23 17 2052-2056
[11]. DOBZHANSKY T 1946 Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura Genetics 31(3) 269–290
[12]. Farmer H McCabe N Lord C J Tutt A N Johnson D A Richardson T B Santarosa M Dillon K J Hickson I Knights C Martin N M Jackson S P Smith G C & Ashworth A 2005 Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy Nature 434(7035) 917–921
[13]. Ferrara R Simionato F Ciccarese C Grego E Cingarlini S Iacovelli R Bria E Tortora G & Melisi D 2018 The development of PARP as a successful target for cancer therapy Expert review of anticancer therapy 18(2) 161–175
[14]. Murai J Huang S Y Das B B Renaud A Zhang Y Doroshow J H Ji J Takeda S & Pommier Y 2012 Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors Cancer research 72(21) 5588–5599
[15]. D'Andrea A D 2018 Mechanisms of PARP inhibitor sensitivity and resistance DNA repair 71 172–176
[16]. Patel A G Sarkaria J N & Kaufmann S H 2011 Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells Proceedings of the National Academy of Sciences of the United States of America 108(8) 3406–3411
[17]. Slade D 2020 PARP and PARG inhibitors in cancer treatment Genes Dev 345-6 360-394 Ferraris D V 2010 Evolution of polyADP-ribose polymerase-1 PARP-1 inhibitors From concept to clinic J Med Chem 5312 4561-4584
[18]. Purnell M R & Whish W J 1980 Novel inhibitors of poly(ADP-ribose) synthetase The Biochemical journal 185(3) 775–777
[19]. Ferraris D V 2010 Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors From concept to clinic J Med Chem 53(12) 4561-4584
[20]. Benchuan C 2015 olaparib a new drug for the treatment of advanced ovarian cancer Medical Review 34 06 846-849