







1. Introduction
The global aged population (65 years old or older) reached 9% in 2020, and this number is expected to increase two-fold in 2050.[1] As life expectancy continues to rise and as aged population continues to enlarge, more people become suffering from age-related diseases while becoming older.[2] Compared to considering the lifespan, people should focus more on healthspan, the time period they live in good health. However, only few studies revealed a corner of the healthspan. To balance between healthy body function and the delay of aging process, it is crucial to discover aging and its related mechanisms, and to develop treatments to extend healthspan.
Aging and lifespan are highly regulated by genetic factors in humans. Large-scale genome-wide association studies (GWAS) have recently identified multiple loci that influence key human aging pathways, and found that there is an association between parental and offspring lifespan.[3] Many genetic diseases that severely affect lifespan, such as progeria, are also hereditary.[3] In the model organism for studying aging, Caenorhabditis elegans, several genes are found to be correlated with nematode lifespan. For example, a mutation on daf-2 or age-1 gene can dramatically extend lifespan in C. elegans.[4] Another disease that highly relates to aging is cancer, which shares similar signaling and regulating pathways to aging. Some cancer-related genes are also lifespan-extending genes, such as the TOR (Target Of Rapamycin) pathway.[5] Firstly discovered in yeast, this highly conserved pathway regulates multiple eukaryotic functions, including aging and cancer.[5] Genetic inhibition of the TORC1 pathway has been shown to extend lifespan and inhibit cancer in C. elegans.[5] However, its function on healthspan still remains mostly unknown.
In this study, C. elegans was used as the model organism. C. elegans has been widely employed as an animal model in aging research due to various advantages, foremost being the ease of synchronization on life stage and short life cycle. In addition, C. elegans is the first multicellular eukaryotic organism to have its complete genome being sequenced, making it a powerful model organism in genetic researches.[6] Many lifespan-regulating genes found in C. elegans are evolutionarily conserved. For example, let-363 in nematode is the ortholog of human gene mTor.[7] Multiple pathways that regulate aging were also experimentally testified in C. elegans, including the TOR signaling pathway, which regulates nematode lifespan by inhibiting autophagy and by promoting the accumulation of oxidative stress.[4,6,8,9,10] The proteins in TOR pathway are also highly evolutionarily conserved.[11]
In this study, RNA interference assay was performed to identify the effect of TOR complex-related genes on lifespan. The online STRING database was used to identify the predicted interactomes of TOR pathway, including let-363 (ortholog of human gene mTor[7]) interactome genes (F56F11.4, ppfr-4, epg-9, aakg-1, lmp-1, rho-1, and ddb-1), daf-15 (ortholog of human gene Rptor[7]) interactome genes (lmtr-2, lmtr-3, atg-13, pitp-1, and F16A11.1), rict-1 (ortholog of human gene rictor[7]) interactome genes (tln-1, mek-2, lat-1, tns-1, and cyd-1), and sinh-1 (ortholog of human gene Mapkap1[7]) interactome genes (mlst-8, R04A9.7, ckc-1, and R10H10.7). Lifespan-shortening genes were already well-studied[12-14], but not enough attention was paid to genes that extend lifespan. Therefore, we decided to focus on lifespan-extending genes after RNAi screening. After identifying F56F11.4 as the lifespan-extending gene, we applied double knockdown assay and RT-qPCR assay to examine the regulatory relationships between the identified gene F56F11.4 and its interactome let-363.
The genetic studies on aging could possibly pinpoint new drug targets to increase human lifespan and healthspan. Therefore, this study may provide new insights intohuman aging and contribute to new anti-aging drug designs.
2. Materials and Methods
2.1. Nematode Strains and Synchronization
All C. elegans strains were incubated at 22.5°C on NGM media seeded with OP50 E. coli as described by Stiernagle.[15] N2 was used as the wild-type strain and synchronized to eggs by bleaching.
2.2. RNA Interference Assay
Bacterial feeding RNAi experiments were performed as described by Kamath et al., and the RNAi clones were derived from the Ahringer RNAi library.[16] Twenty genes related to TOR pathway were included (cyd-1, mlst-8, tns-1, ncbp-2, F16A11.1, lat-1, atg-13, ppfr-4, pitp-1, lmp-1, lmtr-2, F56F11.4, mek-2, R04A9.7, ckc-1, ddb-1, lpin-1, rho-1, lmtr-3, and aakg-1). The bli-3 RNAi clone was set as positive control group for RNAi assay (Figure 1), the age-1 RNAi clone was set as positive control group for lifespan-extending phenotype, and the daf-16 RNAi clone was set as positive control group for lifespan-shortening phenotype, while pL4440 (EV) was the empty control group. The synchronized worms were subjected to RNAi bacteria from L4 stage, which were then used to identify the effect of RNAi on aging phenotypes, including lifespan and pharynx pumping rate. All RNAi clones were confirmed by comparing the sequencing results with NCBI-Blast, and no significant off-target was predicted.

Figure 1. A group with bli-3 RNAi was carried out along with each experiment to test the effectiveness of RNAi treatment. At D3 stage, blisters were found at the surface of treated worms. Scale bar = 0.5 mm.
2.3. Lifespan Assay
For all lifespan assays in this study, age-synchronized L4 larvae were manually transferred to NGM plates, and seeded with E. coli with RNAi plasmids (N = 40). Worms were kept at 22.5°C and scored every two days as dead or alive based on their response to a gentle touch with a wire. Worms that presented externalization of internal organs, died because of bagging, or crawled up the wall of the dish were censored.
2.4. Pharynx Pumping Rate Assay
Pumping rate was monitored and recorded by filming worms (N = 10) at the indicated time point (D1, D5, D9, and D13 stages). Recordings of 25-30 seconds were obtained on Laika M165 microscope using the camera of iPhone 13 Pro Max. One pharyngeal contraction was defined as a cycle of backward and forward movements, and the number of contractions were accurately counted during a 20-second interval. For each strain, the pumping rate is calculated as pumping rate = average N / min.
2.5. RT-qPCR
Synchronized eggs were placed on NGM plates seeded with RNAi bacteria. The worms grew from hatching until day one of adulthood unless otherwise indicated. The worm samples were then harvested and washed with M9 buffer to remove bacteria from the samples. To separate RNA from protein and other materials, worm samples underwent centrifugation in a centrifuge. RNA was then extracted and cDNA was generated by reverse transcription of the total RNA samples. qPCR was performed and normalized to the levels of tba-1 cDNA, with specified experiment settings in the following table (Table 1). The primer sequences for each gene are listed below (Table 2).
Table 1. Experiment setting in RT-qPCR
Expression level | pL4440 | F56F11.4 mutant | let-363 mutant |
Tested genes | tba-1 | ||
F56F11.4 | |||
let-363 | |||
Table 2. Primer sequences
Gene | Forward primer sequence | Reverse primer sequence |
tba-1 | 5’-GTTCCATCCAGAACAGATGAT CACC-3’ | 5’-CAGCTTCGACTTCTTTCCATAGTCA AC-3’ |
let-363 | 5’-CGCACTTCTGTGGAGTCGAC-3’ | 5’-GAATTGCTTCATCTTTGTGATCATC CTGATAC-3’ |
F56F11.4 | 5’-CGACGTGGATAAATCCATTGA TATCAACTC-3’ | 5’-CCAACCATCTCGTATGTTGAATCAG G-3’ |
2.6. Phylogenetic Tree
Phylogenetic tree was made by Neighbor-joining method in MEGA7. The detailed parameters are: Test of Phylogeny-Bootstrap method; No. of Bootstrap Replications-1000; Substitution Type-Nucleotide; Model/Method-Maximum Composite Likelihood; Substitutions to Included: Transitions + Transversions; Rates among sites-Uniform rates; Pattern among Lineages-Same (Homogeneous); Gaps/Missing Data Treatment-complete deletion.
2.7. Use of Open-Source Online Databases
We utilized the STRING database to predict the interactomes of genes let-363, daf-15, rict-1, and sinh-1 by inputting their respective identifiers and selecting C. elegans as our organism of interest, with default confidence scores set to medium for a comprehensive view of potential interactions. NCBI-Blast was also employed to ensure the specificity of designed primer and RNAi sequences.
2.8. Statistic Analysis
Descriptive statistics for continuous variables are presented as mean ± SEM, whereas categorical variables are presented as percentages. Hazard ratios (HRs) and 95% CIs were calculated by Log-rank model to assess the association between groups.
All analyses were performed using Graphpad Prism software (version 9.5). All P-values for the tests were two-sided and P-values < 0.05 were considered statistically significant.
3. Results
3.1. Deficiency of F56F11.4 shortened lifespan in C. elegans
In lifespan assay, the EV, daf-16, and age-1 controls had results consistent with former studies at 22.5°C.[12,17] Among 20 RNAi groups tested, 14 of them were significantly different from the EV control group. The only RNAi group with the phenotype of shortened lifespan was F56F11.4 (P-value < 0.01, Hazard ratio = 1.726) (Figure 2).
RNAi groups with phenotype of extended lifespan included atg-13 (P-value < 0.01, Hazard ratio = 0.6023), cyd-1 (P-value < 0.05, Hazard ratio = 0.7189), tns-1 (P-value < 0.0001, Hazard ratio = 0.4841), F16A11.1 (P-value < 0.0001, Hazard ratio = 0.3178), lat-1 (P-value < 0.01, Hazard ratio = 0.6306), ppfr-4 (P-value < 0.05, Hazard ratio = 0.7038), pitp-1 (P-value < 0.05, Hazard ratio = 0.6567), mek-2 (P-value < 0.001, Hazard ratio = 0.5694), R04A9.7 (P-value < 0.0001, Hazard ratio = 0.4723), ckc-1 (P-value < 0.0001, Hazard ratio = 0.4009), ddb-1 (P-value < 0.01, Hazard ratio = 0.6431), rho-1 (P-value < 0.0001, Hazard ratio = 0.3664), and lmtr-3 (P-value < 0.05, Hazard ratio = 0.6990) (Supplementary Figure 1). Non-significant groups included ncbp-2 (P-value = 0.1869), mlst-8 (P-value = 0.0539), lmp-1 (P-value = 0.5491), lmtr-2 (P-value = 0.2644), lpin-1 (P-value = 0.1750), and aakg-1 (P-value = 0.5526) (Supplementary Figure 2).
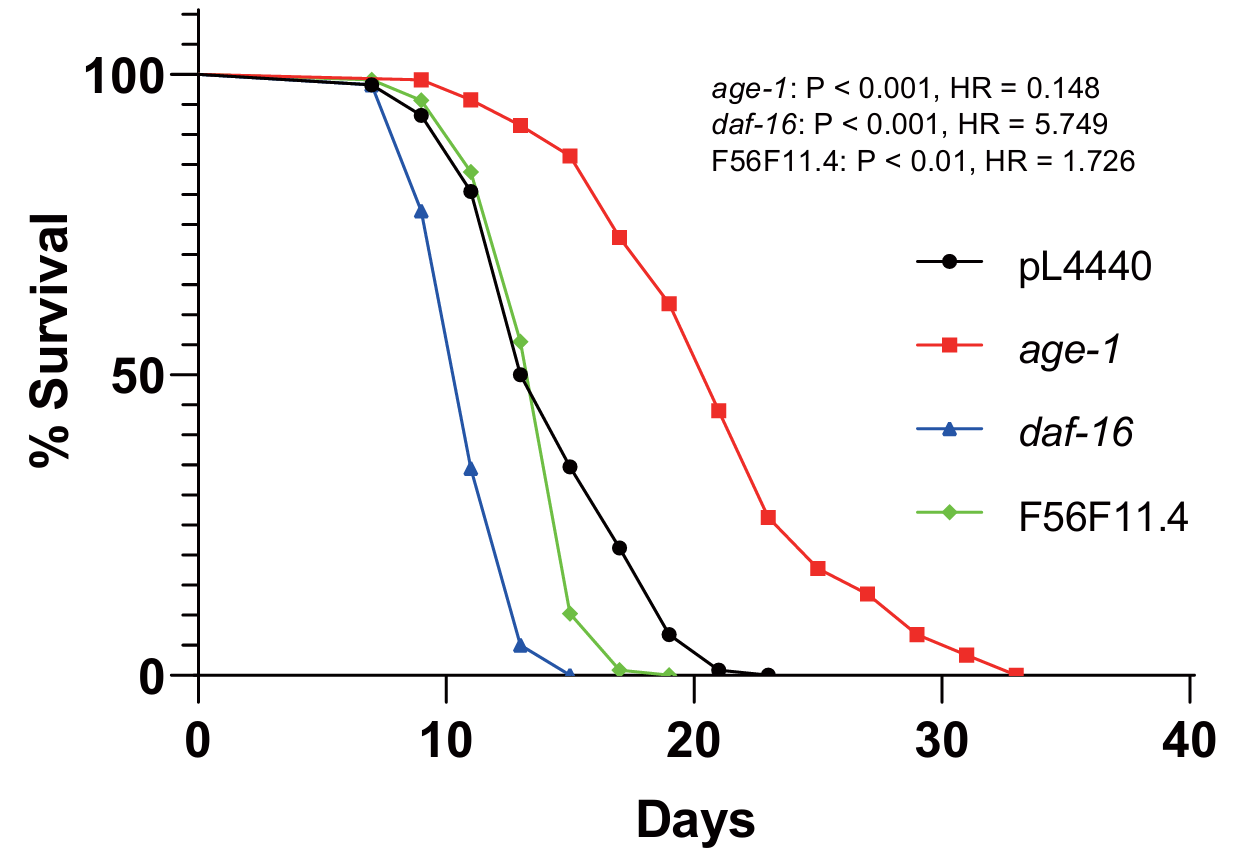
Figure 2. The effect of F56F11.4 knockdown on nematode lifespan. Survival of C. elegans wild-type (black) n = 119, age-1 (red) n = 120, daf-16 (blue) n = 120, and F56F11.4 RNAi (green) n = 120.
3.2. Deficiency of F56F11.4 failed to maintain nematode healthspan in early and late adulthood
After pharyngeal pumping rate assay was done on control groups and all 20 RNAi groups, we compared the pumping rate of every RNAi group with pL4440, at D1, D5, D9, and D13 stages, at which the control group was in a healthy state. Among the above stages, D1 was the baseline, D5 represented early healthspan, D9 represented middle healthspan, and D13 represented late healthspan. For our gene of interest, the knockdown of F56F11.4 significantly decreased nematode pumping rate at early and late healthspan (p < 0.001, p = 0.0105, respectively), as shown in Figure 3. This suggests that F56F11.4 is necessary in maintaining the healthspan of C. elegans.
For other genes, deficiencies of lmtr-2, lat-1, ckc-1, rho-1, lmtr-3, ncbp-2, and lmp-1 all have effect on pumping rate at different periods of adulthood (Supplementary Table 1).
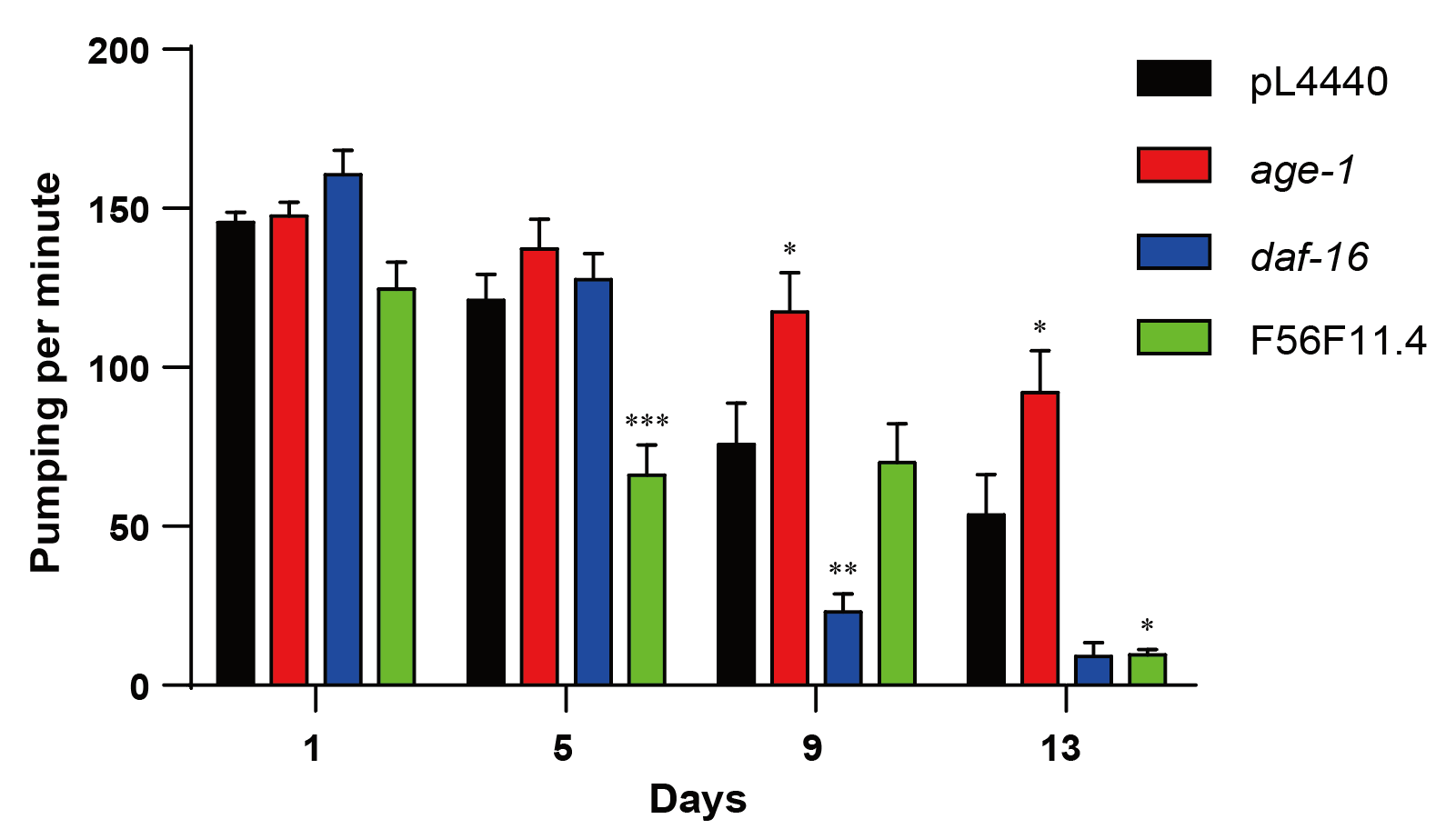
Figure 3. The effect of F56F11.4 knockdown on nematode pumping rate. The pharyngeal pumping rate (average number of contractions per minute) of WT (black), age-1 (red), daf-16 (blue), and F56F11.4 (green) RNAi were recorded on days 1, 5, 9 and 13 of adulthood. *P < 0.05, **P < 0.01, ***P < 0.001.
3.3. Double knockdown of F56F11.4 and let-363 balanced out the phenotype
To examine the regulatory relationships between F56F11.4 and let-363, we used the double knockdown assay to test the two genes. The results of F56F11.4 and let-363 knockdown were consistent with the former single knockdown experiment in this study and previous studies (Figure 4).[12] The deficiencies of both let-363 and F56F11.4 genes contributed to a lifespan longer than F56F11.4 single-knockdown (P-value < 0.001, Hazard ratio = 0.3512) but shorter than let-363 single-knockdown (P-value < 0.001, Hazard ratio = 1.960), as shown in Figure 4.
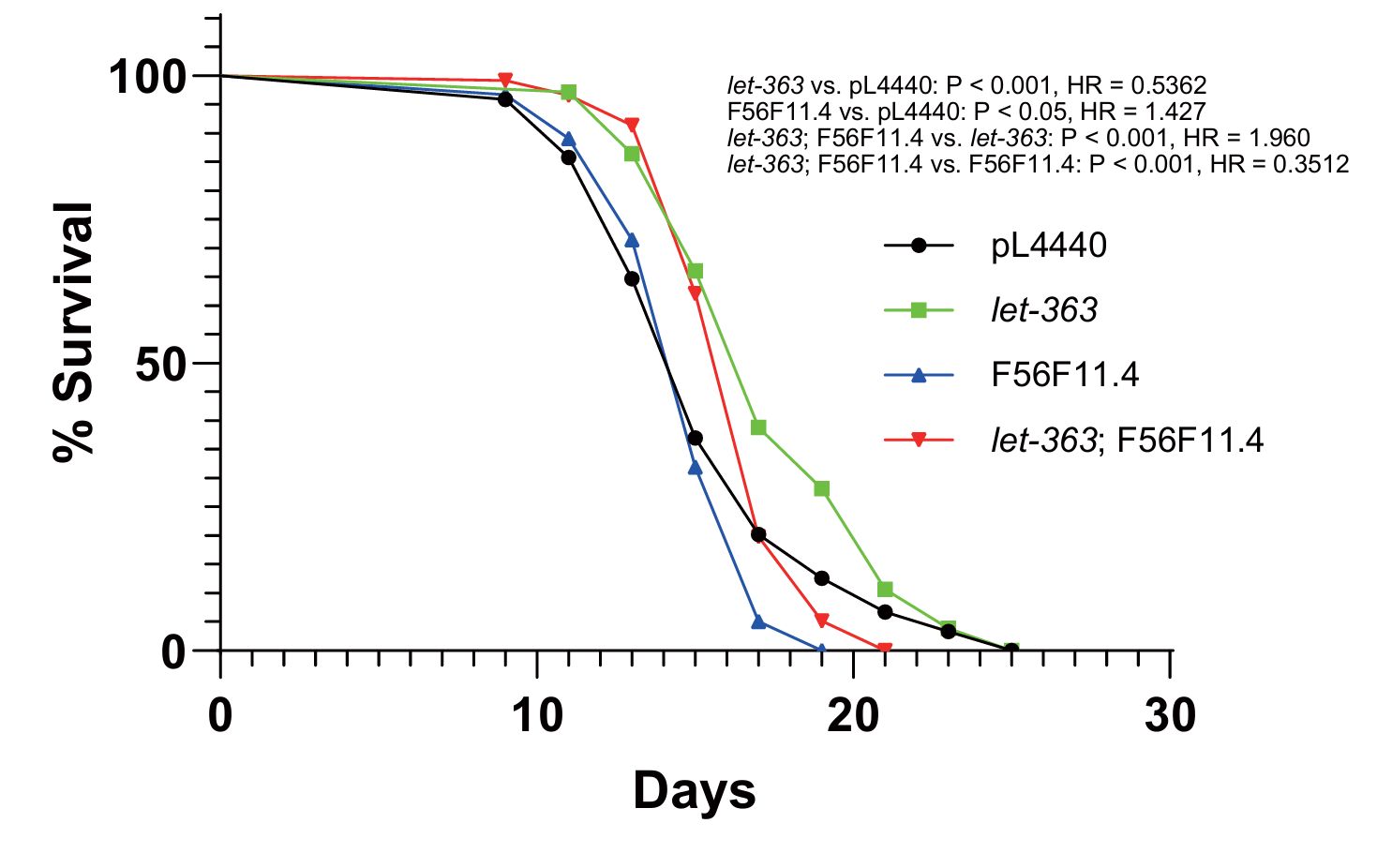
Figure 4. Effect of let-363; F56F11.4 double knockdown on nematode lifespan. Survival of C. elegans wild-type (black) n = 119, let-363 (green) n = 120, F56F11.4 (blue) n = 103, and let-363; F56F11.4 RNAi (red) n = 116.
3.4. Interaction between F56F11.4 and let-363 regulates nematode lifespan
To analyze the regulatory mechanisms between F56F11.4 and let-363, we used RT-qPCR to monitor the expression level of the two genes when one was knocked down. tba-1 was chosen as a constitutively expressed gene for normalization. As shown in Figure 5, when F56F11.4 was subject to RNAi, it showed an 18% reduction in expression level; when let-363 was subject to RNAi, it showed a 53% reduction in expression level. When let-363 was knocked-down, the expression level of F56F11.4 increased 40%; when F56F11.4 was knocked-down, on the contrary, the expression level of let-363 decreased 40% (Figure 5). The 18% knockdown efficiency of F45F11.4 RNAi was enough to increase let-363 expression by 40%, showing that F56F11.4 is a crucial gene, whose little change could lead to a huge alteration in other genes’ expression levels.
When combining the results of RT-qPCR with the previous double knockdown experiment, we inferred about the direction of regulation between let-363 and F56F11.4. Since they have different phenotypes on survival when knocked down, one gene should be inhibiting another if they interact directly. Because let-363 RNAi increased F56F11.4 expression, let-363 should be the upstream inhibitor of F56F11.4.
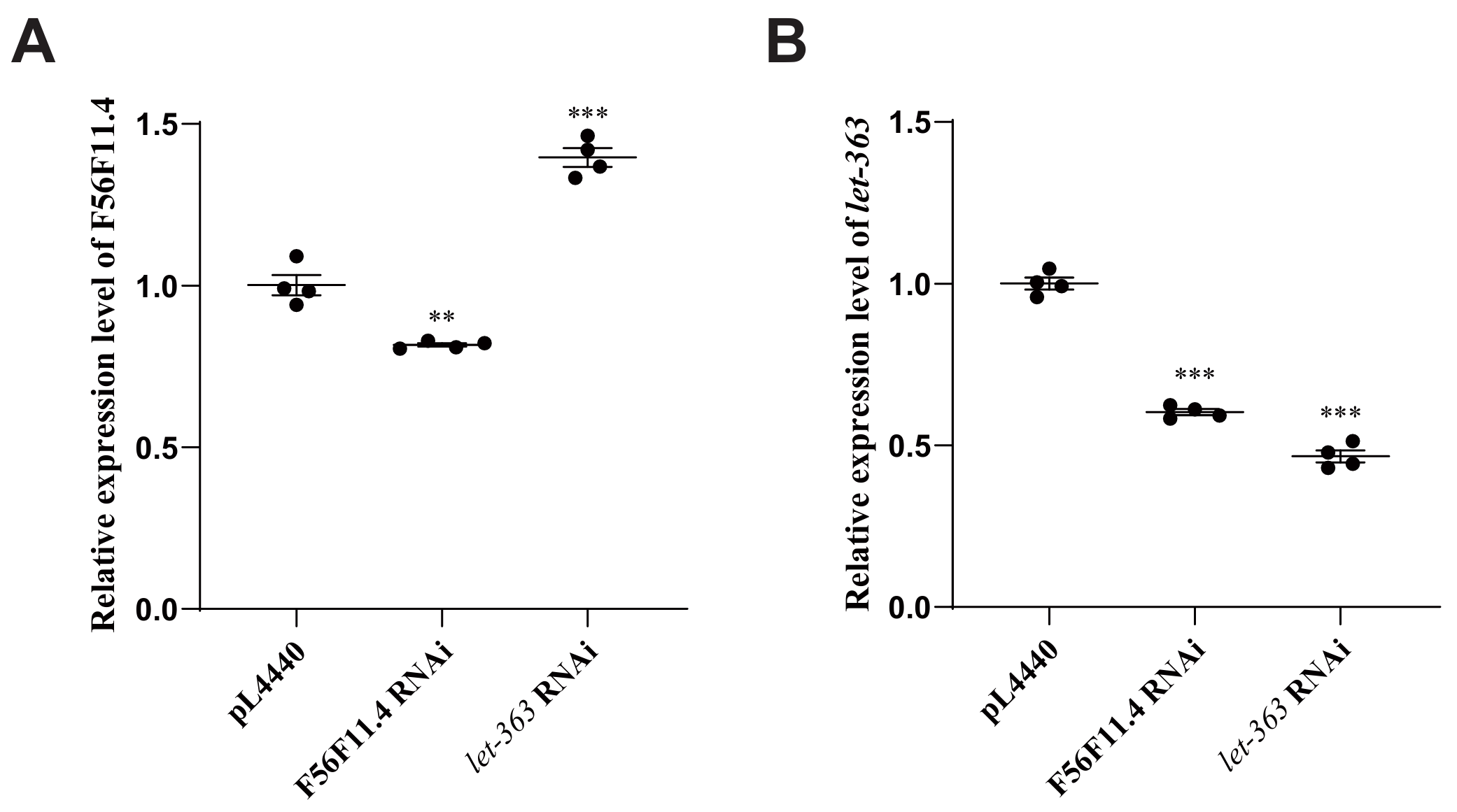
Figure 5. RT-qPCR of F56F11.4 and let-363 expression levels. A, Relative expression level of F56F11.4. B, Relative expression level of let-363. *P < 0.05, **P < 0.01, ***P < 0.001.
3.5. F56F11.4 and let-363 are highly conserved lifespan regulators
To explore the potential of F56F11.4 in human drug design, we then analyzed the homology of C. elegans genes let-363 and F56F11.4 in other organisms, and a phylogenetic tree was made for each gene (Figure 6).
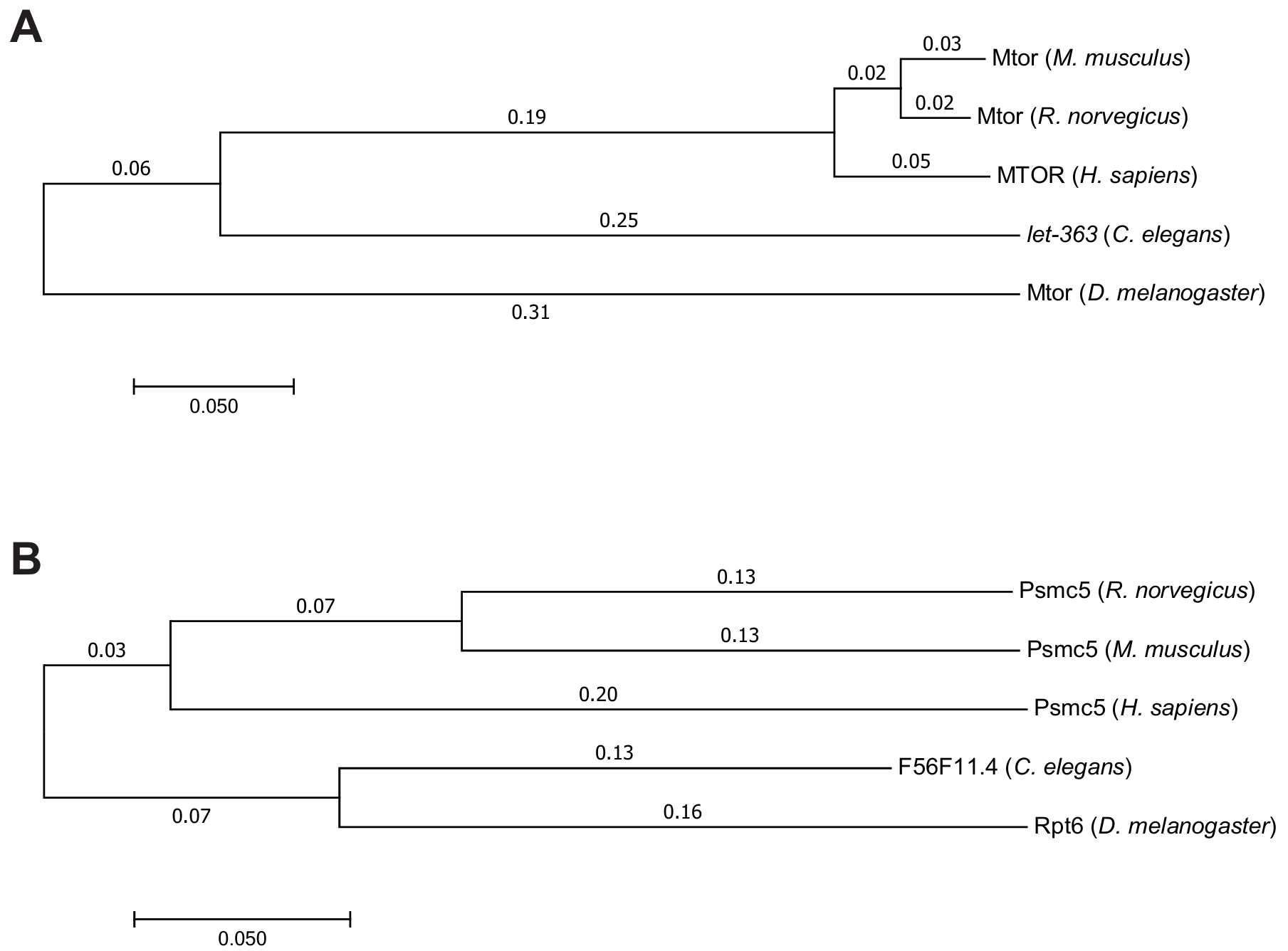
Figure 6. Homologs of C. elegans genes let-363 (A) and F56F11.4 (B) in H. sapiens, M. musculus, R. norvegicus, and D. melanogaster.
For both genes, the evolutionary relationships between H. sapiens, M. musculus, and R. norvegicus were identified to be the closest among all 5 homologs, as those 3 species are all mammals. However, the C. elegans gene let-363 is evolutionarily closer to the mammals’ homologs than to the homolog of D. melanogaster, while F56F11.4 is evolutionarily closer to the homolog of D. melanogaster than to the mammals’ homologs. This might be because mTOR complex and related genes (including let-363) are more evolutionarily conserved than F56F11.4. [8]
4. Discussion
In this work, we aimed to identify the lifespan-extending gene and analyze its mechanism in regulating lifespan. Lifespan assay and pumping rate assay showed that the deficiency of gene F56F11.4 decreased nematode lifespan and healthspan. Further, double knockdown and RT-qPCR uncovered a more precise interaction between this gene and TOR signaling pathway.
4.1. Lifespan is regulated by the conserved TOR pathway
Our experiment showed that the deficiency of let-363, the ortholog of human mTor in C. elegans, extends nematode lifespan, and that this gene is highly evolutionarily conserved among many species, including humans.
In mammals, mTOR functions in regulation of lifespan. Previous experiments in mice, marmosets, and dogs all reported statistically significant results in lifespan or healthspan when treated with rapamycin, an inhibitor of mTOR.[18-20] In humans, due to the moral concerns, there were only clinical trials for small doses of rapamycin or similar mTOR inhibitors.[20] Nonetheless, previous researches have found that those drugs can slow down age-related conditions in multiple organs, including brain, heart, muscle, skin, etc.[20]
The mechanism of TOR pathway regulating lifespan and aging is rather complex. Previous studies reported that TOR shortens yeast lifespan through increasing mitochondrial electron transport chain activity and therefore promoting the accumulation of oxidative stress.[6,9] Another downstream pathway of TOR complex is autophagy, especially mitophagy, the type of autophagy that targets mitochondrion.[21] In this pathway, HLH-30 is the transcription factor regulated by let-363 in C. elegans, and its activated form can increase the expression of downstream autophagy-related genes.[6,10]
4.2. Mechanism of F56F11.4 and rpt-/rpn- families extending lifespan in C. elegans and other species
In identifying the exact mechanism of gene F56F11.4 affecting lifespan, we used the STRING database to look for interactomes of this gene. Among its top 20 predicted interactomes, there are 4 (rpt-1, rpt-2, rpt-3, and rpt-5) in rpt- family and 8 (rpn-1, rpn-3, rpn-5, rpn-6.2, rpn-7, rpn-10, rpn-11, and rpn-13) in rpn- family.
The rpt- and rpn- families both belong to the components of the proteasomal complex. The proteasome maintains cellular homeostasis by degrading oxidized and damaged proteins, a process known to slow down the progress of aging.[22] A former study reveals that upregulation of rpn-6 expression can directly increase lifespan by mitigating oxidative pressure, and this is one of the pathways in which DAF-16/FOXO regulates lifespan.[23] In another study, 4 genes from rpt- and rpn- families were tested and compared on their effect on lifespan, and the deficiency of every tested gene resulted in lifespan reduction.[24]
The rpt- and rpn- families are also closely related to let-363/mTOR pathway. Starvation and inactivation of mTOR complex were previously identified to enhance the activity of autophagy and proteasomal complexes, in which the rpt- and rpn- families locate and function.[25]
Apart from their effect on nematode lifespan, rpt- and rpn- families also regulate aging in other model organisms. In Saccharomyces cerevisiae, the increased expression level of protein Rpn4 can result in increased replicative lifespan.[26] In Drosophila melanogaster, overexpression of Rpn11 also upregulates the lifespan of flies by suppressing aging-dependent reduction in proteasomal activity.[27]
For mammals, there were no previous studies about the direct influence of rpt-/rpn- families on animal aging, but some researches focused on senescence on the cellular level. One study revealed that the high constitutive expression of rpt- and rpn- families contributes to the immortality of embryonic stem cells (ESCs).[28] Mouse ESCs are highly correlated with high expression of Psmd14/Rpn11, a deubiquitinating enzyme in 19S regulatory component of proteasome.[29] Similarly, in human, PSMD11/Rpn6 is highly expressed in hESCs, keeping the cells free from senescence.[30,31]
4.3. The regulatory relationship between TOR pathway and rpt-/rpn- families
In our double knockdown and RT-qPCR experiments, we found that TOR complex and F56F11.4 have a regulatory relationship, in which let-363 inhibits F56F11.4. While this is the first time the interaction between TOR complex and F56F11.4 is identified since F56F11.4 is highly related and interacting with rpt-/rpn- families, past studies had also found that TOR pathway inhibits proteasomal function and activities of rpt-/rpn- families in multiple ways.[32]
First, TOR complex regulates the assembly of regulatory components of proteasome.[33] In yeast, inhibition of TORC1 can cause activation of Adc17, a suppressor for proteasomal dysfunctioning, therefore increasing proteasomal activity.[32,34] Another mechanism is that TORC1 controls proteasome biogenesis, by sensing the signal of the amino acid amount and regulating the catabolic/anabolic pathways.[32] One study used starvation to inhibit TORC1, which led to higher proteasomal function and higher autophagy.[32]
4.4. Lifespan-shortening genes and their mechanisms
Lifespan-shortening genes identified in our knockdown experiment (atg-13, cyd-1, tns-1, F16A11.1, lat-1, ppfr-4, pitp-1, mek-2, R04A9.7, ckc-1, ddb-1, rho-1, and lmtr-3) have different mechanisms in regulating lifespan. The most common way among them is through regulating protein ubiquitination and proteasomal complex, including genes F16A11.1, lat-1, and ddb-1. This is also the pathway in which the identified lifespan-extending gene F56F11.4 regulates lifespan. Since one main function of mTOR complex is inhibiting the proteasome function[11], this result is consistent with previously identified pathways. Another key mechanism of shortening lifespan is through inhibiting autophagy, which was embodied in the gene atg-13, the interactome of daf-15 gene in the mTOR pathway. Since mTOR pathway also has the downstream effect of inhibiting autophagy[8], the position of atg-13 in mTOR pathway was verified.
Other lifespan-shortening genes also use TOR-related pathways to reduce lifespan. They included regulating cell cycle (cyd-1), regulating axon regeneration (tns-1), regulating protein kinase or phosphatase (mek-2, R04A9.7, ckc-1, and ppfr-4), regulating lipid transfer (pitp-1), regulating scaffold protein binding (mek-2), and regulating G protein-coupled receptor (rho-1).[7]
4.5. The value of this study
Potential lifespan-extending drugs are currently being discovered, and many of them are based on TOR pathway. In 2009, rapamycin was verified to extend lifespan in M. musculus, functioning by binding with FKBP12, binding to mTORC1, and inhibiting mTOR pathway.[35] A recently emerged class of senolytic drugs was first discovered in 2015 that could extend lifespan in M. musculus.[36] This type of drug also functions by inhibiting mTOR pathway, and many of those drugs are now in the process of clinical phase 2 trials.[36,37] These drugs demonstrate the potential of aging-related studies based on TOR pathway: to identify new drug targets for clinical use.
5. Conclusion
In this work, we screened out and demonstrated F56F11.4 as a lifespan-extending gene and analyzed its mechanism in regulating lifespan. Double knockdown experiment and RT-qPCR uncovered a more precise regulation between this gene and the TOR signaling pathway. With more and more research being done in the field of human aging, we are optimistic that safe and effective drugs can extend human healthspan in the future.
Acknowledgments
In the course of completing this research, I have faced many challenges, foremost among which being the difficulty of transporting worms to another NGM plate. This process, since I have never done any research using C. elegans before, challenged me a lot in the first few days of my experiments, as I frequently killed the transported worm or broke the gel when I landed, contributing to several failures of my dry lab experiments. To address this problem, I cultured hundreds of worms on a plate and used them to practice by transporting them to another plate for a week. Finally, the number of censored worms became few after my practice, which made my following survival assay quite successful.
Another problem and time-consuming step in the whole project was processing data, especially video data that recorded the pharyngeal pumping of worms. I set up 23 RNAi groups in total, and each group needed 30 worms to include biological repetition at 4 stages of worm adulthood. Therefore, taking and watching those videos took me months to finish.
Through encountering and solving those problems during research, however, I have gained a lot from this process. Apart from my gradual familiarity with and knowledge about C. elegans as the model organism, I also gained better time management skills and patience for long and boring video processing.
As this research work is about to be completed, I would like to express my deepest gratitude to my supervisors, Mr. Ding and Dr. Dou. During my academic journey, they inspired me on the research topic, guided me with their profound professional knowledge and rigorous academic attitude, and provided valuable advice and encouragement when I encountered difficulties and challenges. The wisdom and generosity of them have had a profound impact on me, and I will always remember their contribution to this work.
I also want to thank my classmate Percy, for his technical help and support after I conceived the idea of using Python to analyze pumping rate data. Your advice from a computer science student’s point of view gave me insights into the resources I could use to learn and the implementation details of such a program. Although I eventually gave up this idea, your guide was important and useful in my exploration on this new analytical method.
My heartfelt gratitude goes out to my family and parents, whose love and support are my strongest backing. Special thanks to my father, who helped me contact a commercial biology lab to conduct my experiment in Beijing, and whose unconditional understanding and encouragement were the driving force for me to keep moving forward.
Finally, I would like to thank lab supervisor Dr. Liu, who had provided me with both resources and assistance. He helped me in the use of lab equipment, tutored me on keeping a lab notebook, and once helped me transport several media cultures from one temperature into another when I was absent. I would like to express my sincere gratitude to him.
Please accept my sincere gratitude to all those who have supported and helped me during this academic journey. Without them my research could never be complete and successful.
References
[1]. Jarzebski, M.P., et al., Ageing and population shrinking: implications for sustainability in the urban century. npj Urban Sustainability, 2021. 1(1): p. 17.
[2]. Murray, C.J.L., et al., Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990–2013: quantifying the epidemiological transition. The Lancet, 2015. 386(10009): p. 2145-2191.
[3]. Melzer, D., L.C. Pilling, and L. Ferrucci, The genetics of human ageing. Nature Reviews Genetics, 2019. 21(2): p. 88-101.
[4]. Shen, P., et al., Caenorhabditis elegans: A Convenient In Vivo Model for Assessing the Impact of Food Bioactive Compounds on Obesity, Aging, and Alzheimer's Disease. Annual Review of Food Science and Technology, 2018. 9(1): p. 1-22.
[5]. Beauchamp, E.M. and L.C. Platanias, The evolution of the TOR pathway and its role in cancer. Oncogene, 2013. 32(34): p. 3923-3932.
[6]. Zhang, S., et al., Caenorhabditis elegans as a Useful Model for Studying Aging Mutations. Frontiers in Endocrinology, 2020. 11.
[7]. Kishore, R., et al., Automated generation of gene summaries at the Alliance of Genome Resources. Database, 2020. 2020: p. baaa037.
[8]. Liu, G.Y. and D.M. Sabatini, mTOR at the nexus of nutrition, growth, ageing and disease. Nature Reviews Molecular Cell Biology, 2020. 21(4): p. 183-203.
[9]. Pan, Y., et al., Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab, 2011. 13(6): p. 668-78.
[10]. Ryu, D., et al., Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med, 2016. 22(8): p. 879-88.
[11]. Blackwell, T.K., et al., TOR Signaling in Caenorhabditis elegans Development, Metabolism, and Aging. Genetics, 2019. 213(2): p. 329-360.
[12]. Matei, I.V., et al., Knock-down of odr-3 and ife-2 additively extends lifespan and healthspan in C. elegans. Aging (Albany NY), 2021. 13(17): p. 21040-21065.
[13]. Liu, Y.J., et al., Mitochondrial translation and dynamics synergistically extend lifespan in C. elegans through HLH-30. Journal of Cell Biology, 2020. 219(6).
[14]. Schwarz, F., et al., Knockdown of Indy/CeNac2 extends Caenorhabditis elegans life span by inducing AMPK/aak-2. Aging (Albany NY), 2015. 7(8): p. 553-67.
[15]. Stiernagle, T., Maintenance of C. elegans. WormBook, 2006.
[16]. Kamath, R.S., et al., Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol, 2001. 2(1): p. Research0002.
[17]. Huang, J., et al., Quantitative phosphoproteomics reveals GTBP-1 regulating C.elegans lifespan at different environmental temperatures. Biochemical and Biophysical Research Communications, 2018. 503(3): p. 1962-1967.
[18]. Sills, A.M., et al., Long-term treatment with the mTOR inhibitor rapamycin has minor effect on clinical laboratory markers in middle-aged marmosets. Am J Primatol, 2019. 81(2): p. e22927.
[19]. Urfer, S.R., et al., A randomized controlled trial to establish effects of short-term rapamycin treatment in 24 middle-aged companion dogs. Geroscience, 2017. 39(2): p. 117-127.
[20]. Mannick, J.B. and D.W. Lamming, Targeting the biology of aging with mTOR inhibitors. Nature Aging, 2023. 3(6): p. 642-660.
[21]. Tullet, J.M.A., et al., The SKN-1/Nrf2 transcription factor can protect against oxidative tress and increase lifespan in C. elegans by distinct mechanisms. Aging Cell, 2017. 16(5): p. 1191-1194.
[22]. Chondrogianni, N., et al., Proteasome activation delays aging in vitro and in vivo. Free Radic Biol Med, 2014. 71: p. 303-320.
[23]. Vilchez, D., et al., RPN-6 determines C. elegans longevity under proteotoxic stress onditions. 2012. 489: p. 263-268.
[24]. Sinha, A. and R. Rae, A functional genomic screen for evolutionarily conserved genes equired for lifespan and immunity in germline-deficient C. elegans. PLoS One, 2014. 9(8): p. e101970.
[25]. Guo, B., et al., Genome-wide screen identifies signaling pathways that regulate autophagy during Caenorhabditis elegans development. EMBO reports, 2014. 15(6): p. 705-713-713.
[26]. Kruegel, U., et al., Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet, 2011. 7(9): p. e1002253.
[27]. Tonoki, A., et al., Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol Cell Biol, 2009. 29(4): p. 1095-106.
[28]. Saez, I. and D. Vilchez, The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr Genomics, 2014. 15(1): p. 38-51.
[29]. Buckley, S.M., et al., Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell, 2012. 11(6): p. 783-98.
[30]. Vilchez, D., et al., Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature, 2012. 489(7415): p. 304-8.
[31]. Assou, S., et al., A gene expression signature shared by human mature oocytes and mbryonic stem cells. BMC Genomics, 2009. 10: p. 10.
[32]. Rousseau, A. and A. Bertolotti, An evolutionarily conserved pathway controls proteasome homeostasis. Nature, 2016. 536(7615): p. 184-9.
[33]. Rousseau, A. and A. Bertolotti, Regulation of proteasome assembly and activity in health and disease. Nature Reviews Molecular Cell Biology, 2018. 19(11): p. 697-712.
[34]. Hanssum, A., et al., An inducible chaperone adapts proteasome assembly to stress. Mol Cell, 2014. 55(4): p. 566-77.
[35]. Harrison, D.E., et al., Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. 2009. 460(7253): p. 392-395.
[36]. Guarente, L., D.A. Sinclair, and G. Kroemer, Human trials exploring anti-aging medicines. Cell Metab, 2024. 36(2): p. 354-376.
[37]. Chaib, S., T. Tchkonia, and J.L. Kirkland, Cellular senescence and senolytics: the path to the clinic. Nat Med, 2022. 28(8): p. 1556-1568.
Cite this article
Wang,K. (2024). F56F11.4 Extends Healthspan by Regulation of TOR-Related Pathway in C. elegans. Theoretical and Natural Science,64,68-80.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Jarzebski, M.P., et al., Ageing and population shrinking: implications for sustainability in the urban century. npj Urban Sustainability, 2021. 1(1): p. 17.
[2]. Murray, C.J.L., et al., Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990–2013: quantifying the epidemiological transition. The Lancet, 2015. 386(10009): p. 2145-2191.
[3]. Melzer, D., L.C. Pilling, and L. Ferrucci, The genetics of human ageing. Nature Reviews Genetics, 2019. 21(2): p. 88-101.
[4]. Shen, P., et al., Caenorhabditis elegans: A Convenient In Vivo Model for Assessing the Impact of Food Bioactive Compounds on Obesity, Aging, and Alzheimer's Disease. Annual Review of Food Science and Technology, 2018. 9(1): p. 1-22.
[5]. Beauchamp, E.M. and L.C. Platanias, The evolution of the TOR pathway and its role in cancer. Oncogene, 2013. 32(34): p. 3923-3932.
[6]. Zhang, S., et al., Caenorhabditis elegans as a Useful Model for Studying Aging Mutations. Frontiers in Endocrinology, 2020. 11.
[7]. Kishore, R., et al., Automated generation of gene summaries at the Alliance of Genome Resources. Database, 2020. 2020: p. baaa037.
[8]. Liu, G.Y. and D.M. Sabatini, mTOR at the nexus of nutrition, growth, ageing and disease. Nature Reviews Molecular Cell Biology, 2020. 21(4): p. 183-203.
[9]. Pan, Y., et al., Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab, 2011. 13(6): p. 668-78.
[10]. Ryu, D., et al., Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med, 2016. 22(8): p. 879-88.
[11]. Blackwell, T.K., et al., TOR Signaling in Caenorhabditis elegans Development, Metabolism, and Aging. Genetics, 2019. 213(2): p. 329-360.
[12]. Matei, I.V., et al., Knock-down of odr-3 and ife-2 additively extends lifespan and healthspan in C. elegans. Aging (Albany NY), 2021. 13(17): p. 21040-21065.
[13]. Liu, Y.J., et al., Mitochondrial translation and dynamics synergistically extend lifespan in C. elegans through HLH-30. Journal of Cell Biology, 2020. 219(6).
[14]. Schwarz, F., et al., Knockdown of Indy/CeNac2 extends Caenorhabditis elegans life span by inducing AMPK/aak-2. Aging (Albany NY), 2015. 7(8): p. 553-67.
[15]. Stiernagle, T., Maintenance of C. elegans. WormBook, 2006.
[16]. Kamath, R.S., et al., Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol, 2001. 2(1): p. Research0002.
[17]. Huang, J., et al., Quantitative phosphoproteomics reveals GTBP-1 regulating C.elegans lifespan at different environmental temperatures. Biochemical and Biophysical Research Communications, 2018. 503(3): p. 1962-1967.
[18]. Sills, A.M., et al., Long-term treatment with the mTOR inhibitor rapamycin has minor effect on clinical laboratory markers in middle-aged marmosets. Am J Primatol, 2019. 81(2): p. e22927.
[19]. Urfer, S.R., et al., A randomized controlled trial to establish effects of short-term rapamycin treatment in 24 middle-aged companion dogs. Geroscience, 2017. 39(2): p. 117-127.
[20]. Mannick, J.B. and D.W. Lamming, Targeting the biology of aging with mTOR inhibitors. Nature Aging, 2023. 3(6): p. 642-660.
[21]. Tullet, J.M.A., et al., The SKN-1/Nrf2 transcription factor can protect against oxidative tress and increase lifespan in C. elegans by distinct mechanisms. Aging Cell, 2017. 16(5): p. 1191-1194.
[22]. Chondrogianni, N., et al., Proteasome activation delays aging in vitro and in vivo. Free Radic Biol Med, 2014. 71: p. 303-320.
[23]. Vilchez, D., et al., RPN-6 determines C. elegans longevity under proteotoxic stress onditions. 2012. 489: p. 263-268.
[24]. Sinha, A. and R. Rae, A functional genomic screen for evolutionarily conserved genes equired for lifespan and immunity in germline-deficient C. elegans. PLoS One, 2014. 9(8): p. e101970.
[25]. Guo, B., et al., Genome-wide screen identifies signaling pathways that regulate autophagy during Caenorhabditis elegans development. EMBO reports, 2014. 15(6): p. 705-713-713.
[26]. Kruegel, U., et al., Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet, 2011. 7(9): p. e1002253.
[27]. Tonoki, A., et al., Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol Cell Biol, 2009. 29(4): p. 1095-106.
[28]. Saez, I. and D. Vilchez, The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr Genomics, 2014. 15(1): p. 38-51.
[29]. Buckley, S.M., et al., Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell, 2012. 11(6): p. 783-98.
[30]. Vilchez, D., et al., Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature, 2012. 489(7415): p. 304-8.
[31]. Assou, S., et al., A gene expression signature shared by human mature oocytes and mbryonic stem cells. BMC Genomics, 2009. 10: p. 10.
[32]. Rousseau, A. and A. Bertolotti, An evolutionarily conserved pathway controls proteasome homeostasis. Nature, 2016. 536(7615): p. 184-9.
[33]. Rousseau, A. and A. Bertolotti, Regulation of proteasome assembly and activity in health and disease. Nature Reviews Molecular Cell Biology, 2018. 19(11): p. 697-712.
[34]. Hanssum, A., et al., An inducible chaperone adapts proteasome assembly to stress. Mol Cell, 2014. 55(4): p. 566-77.
[35]. Harrison, D.E., et al., Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. 2009. 460(7253): p. 392-395.
[36]. Guarente, L., D.A. Sinclair, and G. Kroemer, Human trials exploring anti-aging medicines. Cell Metab, 2024. 36(2): p. 354-376.
[37]. Chaib, S., T. Tchkonia, and J.L. Kirkland, Cellular senescence and senolytics: the path to the clinic. Nat Med, 2022. 28(8): p. 1556-1568.