







1. Introduction
CAR-T cell therapy functions basically by utilizing genetically modified T cells that display specially designed receptors on their surface to identify particular tumour antigens, and once activated, the CAR-modified T cells will attack tumour cells as “living drugs” [1]. CAR-T cell therapy is an additional treatment option for hematological malignancies, in addition to standard treatments like chemotherapy, radiation, and hematopoietic stem cell transplantation (HSCT), which is an unprecedentedly successful immunotherapy, has started a brand new era of treatments of hematological malignancies. CARs, which are synthetic receptors that target native cell surface antigens, do not require HLA expression to be recognized, being remarkably superior to transduced T-cell receptors (TCR) and other immunotherapies. CAR-T cells have demonstrated remarkable success in the therapy of hematological malignancies due to their ability to target a broad range of antigens, including anti-Cluster of Differentiation (CD) 19 CARs and B-cell maturation antigen (BCMA). The Food and Drug Administration (FDA) has approved six CAR-T treatments since 2017 [2]. These therapies are usually administered to patients who have undergone two or more lines of systemic therapy.
Though achieving considerable success, there are limitations involved in CAR-T therapy, including efficacy concerns, safety concerns, and high costs. Problems of efficacy can be relapse happening after remission. Moreover, safety concerns can sometimes be fatal, reflected by the toxicity of cytokine release syndrome (CRS). CRS can be life-threatening, as the cytokines released including IL-6, IL-10, and interferon (IFN)-Y can lead to fever, chills, headache and a series of immune reactions. The management of CRS after CAR T-cell therapy is important, having tocilizumab involved. Tocilizumab is an antibody agianst IL-6 receptor and has limited toxicity itself. IL-6 is a cytokine that is regarded to most likely release macrophages; so by targeting IL-6, the efficacy of CAR-T cells will not be greatly affected with the CRS syndrome managed simultaneously [3].
Furthermore, the cost of CAR-T cell therapy is a significant concern. The complex process, long time to wait due to the personlized feature of the therapy, and most importantly the consequently high cost would hinder patients from receiving the therapy. CAR-T cells might fetch up to 6–8 times the US GDP per capita, or around US$66,100, on the market in the US for between US$350,000 and US$500,000. For Asian countries typically, a huge barrier of patients getting access to CAR-T therapy is the cost, having the concern of large sums of out-of-pocket costs despite the existence of healthcare insurance system. With this in mind, universal CAR-T cells have been explored as a novel kind of CAR-T cell that are likely to mitigate most of the drawbacks of costs [4].
This review will illustrate the present limitations of CAR-T cell therapy in the treatment of hemtalogical malignancies and propose corresponding strategies to overcome the obstacles. This article will also provide novel designs of CAR-T cell therapies that might be extensively explored in the future.
2. Efficacy concerns and strategies to overcome
2.1. Mechanism of antigen loss
As shown in Figure 1, CAR-T cells kill target cells by encountering a level of number of target antigens that CARs are going to bind to on cancer cells, and then get activated to kill them. Cases of antigen loss typically regarding CD19 loss in CAR-T therapy are categorized into three main groups: antigen escape, lineage switch, and antigen downregulation [5]. In cases of antigen escape, patients’ cells fail to express CD19 molecules that can bind to anti-CD19 molecules together with CARs. Turning to lineage switch, it happens when patients relapse with a genetically related but phenotypically distinct malignancy, often occurring in acute myeloid leukemia (AML) [6]. The third group is antigen downregulation, which results in inadequate amount of expression of antigens. Consequently, there is a lack of CAR-T cell activation.
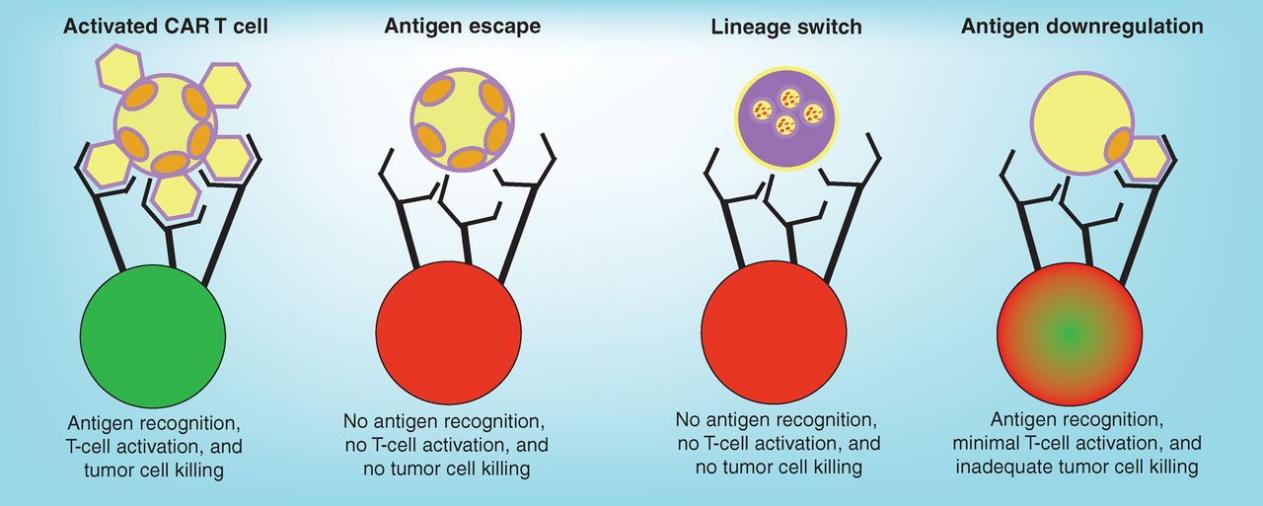
Figure 1. Mechanism of antigen loss [6].
Tumour cells circumvent killing by expressing alternative forms of antigens that cannot bind to CARs (antigen escape), or switching to a genetically related but phenotypically different disease( lineage switch), or by reducing the number of antigens expressed on the surface of cancer cells so that the killing is not complete for all tumour cells (antigen downregulation).
2.2. Strategies to overcome antigen loss
2.2.1. Engineer a T-cell product to target multiple antigens
There are various ways to achieve multispecificity of a CAR-T cell product [6]. The first pattern is to give CAR-T cell products that target different antigens (with different CARs) simultaneously or follow a sequence for administration. Or to simplify this process, there can be novel designs of T-cell products, which not only targeting one antigen, but also are positive for both CARs, achieving a mixed product of treatment. This can consequently achieve amplified function when there are multiple antigens involved. These two ways are called coadministration and cotransduction respectively [7]. However, there are drawbacks to these designs, as extensive production of T-cell products can have high costs of labor, resulting in unaffordable prices.
2.2.2. Novel CAR designs
One CAR molecule can also target several antigens, which can be engineered to link tow binders on one molecule. Eugenia Zah et al, constructed novel designs of CARs that are capable of OR-gate signal processing--CARs that can bind to either CD19 or CD20 whenever each one of them are lack of expression or fail to bind to CARs. In this way, the effects of the killing of tumour cells can be enhanced, as the targeting of malignant B cells in acute and chronic B-cell malignancies can be achieved through targeting either antigen. These CARs are called bispecific CARs, and a concern related to this method is the clinical outcome of immunogenicity of the next-generation CAR molecules, as there are more points where peptides fuse in receptors. Another concern is the way to optimize the structure of the CARs, including the adjustment of extracellular spacer length and linker sequence between two scFv domains, which will lead to good clinical outcomes, not compromising the efficiency of a single targeting of either antigen [8]
2.2.3. Overcoming low level of antigens
The strategies of dealing with an inadequate amount of target antigens are divided into two ways: administering agents to facilitate the expression of target antigens and CAR engineering to simplify its activity against lower antigen densities. The first way, for example, has been proven effective in preclinical studies. Rachel C Lynn et, al and her colleagues had proved all-trans retinoic acid (ATRA) effective in enhancing the expression of FRβ (a target antigen) in the treatment of AML, which can be better recognized by m909 CAR-T cells [9].
CAR designs that can lead to activation of CAR-T cells in a low level of antigen can be achieved by increasing the affinity of the ScFv for its target. Xiaojun Liu et al, and his colleagues have proved the CARs of adjusted affinity in the ScFv region empowering the wider use of CAR-T cells to treat undruggable cases due to low antigen density [10].
3. Safety concerns and strategies to overcome
3.1. CRS
The syndrome is an inflammatory response after CAR-T cell infusion. CRS is brought on by a significant release of inflammatory markers and cytokines, including as ferritin, interferon-Τ, C-reactive protein, and interleukin (IL-6). Fever is one of the warning signs, which can more dangerously develop into heart attack-threatening vasodilatory shock, capillary leak, and hypoxic respiratory failure. However, the duration can be regulated by intervention after 2 to 3 weeks after CAR-T cell infusion [11].
3.2. Strategies to manage CRS
To understand the concerns of the management of CRS, we have to keep in mind that the proliferation and functioning of CAR-T cells with durable remissions are correlated with CRS. Thus controlling CRS might to some extent influence the antitumour ability of CAR-T cells. However, tocilizumab, an antibody blocking the IL-6 receptor, has achieved mediation of CRS while not influencing the normal functioning of T-cells. Tocilizumab is remarkably distinctive because of its ability to mediate the concern of efficacy and safety simultaneously. By contrast, corticosteroids, which may be effective in some cases, can be detrimental to CAR-T cells.
However, there is no standard algorithm to manage CRS, as many potentially effective agents that can block cytokines are under clinical trials-----anakinra, ruxolitinib, etc [12]. Besides, other than blocking certain cytokines, new CAR-T cell products can be designed to incorporate suicide targets to reduce toxicity. For example, Tobias Riet and his colleagues have modified a CAR by RNA transfection and have tested it effective in adoptive immunotherapy [13].
Of course, managing CRS should take various factors into concern, including the specific CAR-T cell product used, and the individual situation of different patients. The progress of mitigating CRS still has large potential, with more therapeutics being explored continuously.
3.3. How to deliver safe CAR-T cell therapy
The safe access of patients to CAR-T cell therapy is significant during the process of treatment. The selection of patients who can receive CAR-T cell therapy is paramount among all factors, taking patients’ diseases other than hematological malignancies into concern. Additionally, the rapid administration of drugs to mediate toxicity of CAR-T cell therapy should be provided to patients after CAR-T cell infusion. Generally, a multidisciplinary endeavor with expertise, collaboration and patients committed to the oversight are all required to deliver CAR-Ts safely [12].
4. CAR-T Cell Therapy in the future
4.1. Universal CAR-T Cell products
UCAR-T cells can be superior to autologous CAR-T cells in several ways. Firstly, its source of T cells is different from autologous CAR-T cell therapy. CAR-T cell therapy sometimes fails because of the difficulty of harvesting T cells from patients that have received chemotherapy or radiotherapy. In addition, the quality and persistence of these T cells may be unwell, hindering the proliferation and functioning of these cells. However, UCAR-T cells avoid this problem, as it extract T cells from healthy donors, increasing the efficacy and reducing the rate of failure. Another advantage of UCAR-T cell therapy is the time and price involved in getting access to CAR-T cells. Due to the potential large-scale production processes, the time-consuming procedure of designing personalized CAR-T cells can be elided, with large amounts of allogeneic CAR-T cells generated from single healthy donor and provided to patients immediately when needed.
However, the safety concern regarding UCAR-T therapy of patients’ immune systems recognizing the cells as non-self can be significant. Consequently, graft rejection and graft-versus-host disease (GVHD) can be extremely severe. Delightfully, this drawback has been explored extensively and several treatments are under preclinical and clinical studies, such as gene-modified conventional T cells and virus-specific T cells [14].
4.2. Next-generation CARs
Although CAR-T cell therapy has shown great success, there are still concerns that cannot be dealt with effectively, leading to scientists’ attention turning to alternative types of cells that can be utilized for CAR cell therapy--NK cells(natural killer cells)and macrophages. These cells have the advantage of not causing GVHD as they attach target cells directly to the innate immune system without MHC. So they can be potential candidates for the production of Universal CARs [15, 16].
NK cells are advantageous as they can directly identify target antigens without the presence of MHC molecules. In this way, a certain extent of antigen escape can be avoided, as they function uninfluenced by the downregulation of MHC molecules. Additionally, induced pluripotent stem cells and other allogeneic sources can be utilized for the extraction of NK cells, as their activation can be achieved without MHCs. NK cells have shown great efficacy in the treatment of hematological malignancies with no significant toxicity being reported [17].
Given that they are the innate immune cell type with the highest rate of infiltration, macrophages may hold promise. The purpose of CAR-M is to enhance macrophage antigen presentation and phagocytic activity in the fight against tumors. The development of CAR-M cells can also express anti-tumour cytokines and secrete MMPs to degrade the extracellular matrix [18].
Various attempts are being made to explore CAR-T cell therapy currently, and a lot of them are already under preclinical studies and clinical trials, giving people new inspirations and thoughts in the meantime.
5. Conclusion
It has been determined that CAR-T cell therapy is incredibly successful in treating hematological malignancies. However, the drawbacks of the therapy have to be paid more attention to. In this review, efficacy issues and safety concerns are discussed. Antigen loss is a main factor of relapse, usually because of the inadequate expression of surface antigens, leading to the failure of targeting the antigens. Methods towards this issue involves novel designs of T-cells and CARs, making them being able to be bispecific. However, there are still some problems that should be dealt specifically when combining the effect of targeting various antigens together, making sure that the combined targeting effect would not be compromised in each specific design. Regarding safety concerns, CRS is the main topic. Using tocilizumab is an effective way to mitigate the syndrome, however; more drugs are developed and trialed to deal with CRS in various cases and ensure the potential of T-cells not be affected. Turning to the reduction of costs., universal CAR-T therapy is extensively discussed. The widely suitable T-cells can be produced in a large amount to make the therapy more affordable. Regarding the potential obstacles involved, NK cells and macrophages can be designed to be CAR-NKs and CAR-Ms to avoid the GVHD and be more efficient in the treatment of hematological malignancies. In this discussion, other factors of relapse are not discussed, including the tumour microenvironment, and the lack of persistence of CAR-T cells. The development of CAR-T cell therapy is going to face more challenges during the wider application of the therapy, thus getting more attention into innovative therapies to mitigate the imperfections and relapses of CAR-T cell therapy. More attention should also be paid to new ways to reduce the costs, making sure that increasing number of patients are able to receive the therapy and being cured finally.
References
[1]. Sadelain, M., Brentjens, R., & Rivière, I. (2013). The basic principles of chimeric antigen receptor design. Cancer Discovery, 3(4), 388–398.
[2]. Zhang, X., Zhu, L., Zhang, H., Chen, S., & Xiao, Y. (2022). CAR-T cell therapy in hematological malignancies: Current opportunities and challenges. Frontiers in Immunology, 13, 927153.
[3]. Lee, D. W., Gardner, R., Porter, D. L., Louis, C. U., Ahmed, N., Jensen, M., ... & Mackall, C. L. (2014). Current concepts in the diagnosis and management of cytokine release syndrome. Blood, 124(2), 188-195.
[4]. Lin, H., Cheng, J., Mu, W., Zhou, J., & Zhu, L. (2021). Advances in universal CAR-T cell therapy. Frontiers in Immunology, 12, 744823.
[5]. Huang, L., Wang, N., Li, C., Cao, Y., Xiao, Y., Xiao, M., et al. (2017). Sequential infusion of anti-CD22 and anti-CD19 chimeric antigen receptor T cells for adult patients with refractory/relapsed B-cell acute lymphoblastic leukemia. Blood, 130, 846.
[6]. Majzner, R. G., & Mackall, C. L. (2018). Tumor antigen escape from CAR T-cell therapy. Cancer Discovery, 8(10), 1219–1226.
[7]. Majzner, R. G., Heitzeneder, S., & Mackall, C. L. (2017). Harnessing the immunotherapy revolution for the treatment of childhood cancers. Cancer Cell, 31, 476–485.
[8]. Zah, E., Lin, M.-Y., Silva-Benedict, A., Jensen, M. C., & Chen, Y. Y. (2016). T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunology Research, 4(6), 498–508.
[9]. Lynn, R. C., Poussin, M., Kalota, A., Feng, Y., Low, P. S., Dimitrov, D. S., & Powell Jr., D. J. (2015). Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood, 125(22), 3466-3476.
[10]. Liu, X., Jiang, S., Fang, C., Yang, S., Olalere, D., Pequignot, E. C., ... & Zhao, Y. (2015). Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Research, 75(17), 3596-3607.
[11]. Frey, N. V., & Porter, D. L. (2016). Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. In Hematology: American Society of Hematology Education Program Book (Vol. 2016(1), pp. 567–572).
[12]. Frey, N., & Porter, D. (2019). Cytokine release syndrome with chimeric antigen receptor T cell therapy. Biology of Blood and Marrow Transplantation, 25(4).
[13]. Riet, T., Holzinger, A., Dörrie J., Schaft N., Schuler G., & Abken H. (2013). Nonviral RNA transfection to transiently modify T cells with chimeric antigen receptors for adoptive therapy. In Methods in Molecular Biology (Vol. 969, pp. 187-201).
[14]. Leen A.M., Bollard C.M., Mendizabal A.M., Shpall E.J., Szabolcs P., Antin J.H., ... & Kapoor N.(2013). Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation.Blood, 121, 5113–5123.
[15]. Zhao J., Lin Q., Song Y., & Liu D.(2018). Universal CARs, universal T cells and universal CAR T cells.Journal of Hematology & Oncology, 11, 1-9.
[16]. Chen Y.J., Abila B, . & Mostafa Kamel Y.(2023). CAR-T: What is next?Cancers (Basel), 15(3), 663.
[17]. Chen Y, . Yu Z, . Tan X, . Jiang H, . Xu Z, . Fang Y, . Han D, . Hong W, . Wei W, . & Tu J.(2021). CAR-macrophage: A new immunotherapy candidate against solid tumors.Biomedicine & Pharmacotherapy, 139, 111605.
[18]. Liu E, . Marin D, . Banerjee P, . Macapinlac H.A, . Thompson P, . Basar R, . Nassif Kerbauy L, . Overman B, . Thall P, . Kaplan M, . et al.(2020). Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors.New England Journal of Medicine, 382, 545–553.
Cite this article
Kang,E. (2024). Current status and optimization of CAR-T therapy in hematological malignancies. Theoretical and Natural Science,67,120-125.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Sadelain, M., Brentjens, R., & Rivière, I. (2013). The basic principles of chimeric antigen receptor design. Cancer Discovery, 3(4), 388–398.
[2]. Zhang, X., Zhu, L., Zhang, H., Chen, S., & Xiao, Y. (2022). CAR-T cell therapy in hematological malignancies: Current opportunities and challenges. Frontiers in Immunology, 13, 927153.
[3]. Lee, D. W., Gardner, R., Porter, D. L., Louis, C. U., Ahmed, N., Jensen, M., ... & Mackall, C. L. (2014). Current concepts in the diagnosis and management of cytokine release syndrome. Blood, 124(2), 188-195.
[4]. Lin, H., Cheng, J., Mu, W., Zhou, J., & Zhu, L. (2021). Advances in universal CAR-T cell therapy. Frontiers in Immunology, 12, 744823.
[5]. Huang, L., Wang, N., Li, C., Cao, Y., Xiao, Y., Xiao, M., et al. (2017). Sequential infusion of anti-CD22 and anti-CD19 chimeric antigen receptor T cells for adult patients with refractory/relapsed B-cell acute lymphoblastic leukemia. Blood, 130, 846.
[6]. Majzner, R. G., & Mackall, C. L. (2018). Tumor antigen escape from CAR T-cell therapy. Cancer Discovery, 8(10), 1219–1226.
[7]. Majzner, R. G., Heitzeneder, S., & Mackall, C. L. (2017). Harnessing the immunotherapy revolution for the treatment of childhood cancers. Cancer Cell, 31, 476–485.
[8]. Zah, E., Lin, M.-Y., Silva-Benedict, A., Jensen, M. C., & Chen, Y. Y. (2016). T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunology Research, 4(6), 498–508.
[9]. Lynn, R. C., Poussin, M., Kalota, A., Feng, Y., Low, P. S., Dimitrov, D. S., & Powell Jr., D. J. (2015). Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood, 125(22), 3466-3476.
[10]. Liu, X., Jiang, S., Fang, C., Yang, S., Olalere, D., Pequignot, E. C., ... & Zhao, Y. (2015). Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Research, 75(17), 3596-3607.
[11]. Frey, N. V., & Porter, D. L. (2016). Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. In Hematology: American Society of Hematology Education Program Book (Vol. 2016(1), pp. 567–572).
[12]. Frey, N., & Porter, D. (2019). Cytokine release syndrome with chimeric antigen receptor T cell therapy. Biology of Blood and Marrow Transplantation, 25(4).
[13]. Riet, T., Holzinger, A., Dörrie J., Schaft N., Schuler G., & Abken H. (2013). Nonviral RNA transfection to transiently modify T cells with chimeric antigen receptors for adoptive therapy. In Methods in Molecular Biology (Vol. 969, pp. 187-201).
[14]. Leen A.M., Bollard C.M., Mendizabal A.M., Shpall E.J., Szabolcs P., Antin J.H., ... & Kapoor N.(2013). Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation.Blood, 121, 5113–5123.
[15]. Zhao J., Lin Q., Song Y., & Liu D.(2018). Universal CARs, universal T cells and universal CAR T cells.Journal of Hematology & Oncology, 11, 1-9.
[16]. Chen Y.J., Abila B, . & Mostafa Kamel Y.(2023). CAR-T: What is next?Cancers (Basel), 15(3), 663.
[17]. Chen Y, . Yu Z, . Tan X, . Jiang H, . Xu Z, . Fang Y, . Han D, . Hong W, . Wei W, . & Tu J.(2021). CAR-macrophage: A new immunotherapy candidate against solid tumors.Biomedicine & Pharmacotherapy, 139, 111605.
[18]. Liu E, . Marin D, . Banerjee P, . Macapinlac H.A, . Thompson P, . Basar R, . Nassif Kerbauy L, . Overman B, . Thall P, . Kaplan M, . et al.(2020). Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors.New England Journal of Medicine, 382, 545–553.