







1. Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder that begins with mild memory loss and progresses to language difficulties, disorientation, impaired judgment, and emotional changes[1,2]. The disease, initially reported by German physician Alois Alzheimer in 1906 and named after him, is now commonly known as Alzheimer's disease [12/24/2024 4:54:00 PM3]. AD is the most typical form of dementia, accounting for 60-80% of cases in the elderly group [4]. As the disease progresses, patients may ultimately lose the ability to live independently, requiring around-the-clock care [1,5]. Alzheimer's disease is currently incurable, and existing treatments centre on symptom relief and progression delay [2,6,7]. The disease's specific onset mechanisms are unclear, but genetics, environmental factors and lifestyle all significantly contribute.
In AD, β-amyloid peptide (Aβ) is generated from amyloid precursor protein (APP) by β and γ secretases. APP is normally processed through the α secretase pathway, not producing Aβ. However, in AD patients, APP is more processed through the β secretase pathway, producing Aβ fragments that accumulate in the brain as amyloid plaques [1,6,7]. The plaques accumulate between neurons, interfering with signal transmission and leading to neuronal death and brain atrophy [1]. Tau protein, a microtubule-associated protein, normally helps maintain cell stability [8]. In AD patients, tau abnormally phosphorylates, detaching from microtubules and forming neurofibrillary tangles [9,10]. These tangles accumulate within neurons, interfering with cellular transport and ultimately leading to neuronal death [11].
Recent studies find a connection between Alzheimer's disease and endoplasmic reticulum (ER) stress due to the accumulation of misfolded or unfolded proteins [12-14]. The unfolded protein response (UPR) is activated to restore normal ER function [15], but if the stress is too strong, cell apoptosis may result. However, when the stress is too intense or prolonged, the UPR cannot restore endoplasmic reticulum homeostasis, leading the cell to initiate apoptosis [16,17]. The build-up of Aβ and abnormal tau protein phosphorylation can trigger ER stress, while continued ER stress worsens their misfolding and accumulation [13,18]. This review focuses on the molecular mechanisms of Alzheimer's disease and the endoplasmic reticulum stress response, exploring the pathways linking them. It aims to provide insight into the underlying molecular mechanisms of Alzheimer's and potential treatments.
2. Pathological Features of AD
2.1. Amyloid Beta Deposition
Amyloid Beta (Aβ) is a key feature of Alzheimer's disease (AD), characterized by Aβ plaque buildup in the brain [1,19]. Aβ and tau proteins can accumulate in amyloid plaques and neurofibrillary tangles, linked to neurodegenerative processes in AD patients' brains [6,20]12/24/2024 4:54:00 PM. Studies have found that mutations in the PSEN1, PSEN2, or APP genes in familial Alzheimer's patients can lead to abnormal processing and aggregation of Aβ proteins [21]. The APP gene precursors Aβ peptides, with mutations affecting cleavage and aggregation. The PSEN1 and PSEN2 genes provide catalytic subunits for the γ-secretase enzyme, breaking down APP to generate Aβ peptides (Figure1) [19,22].
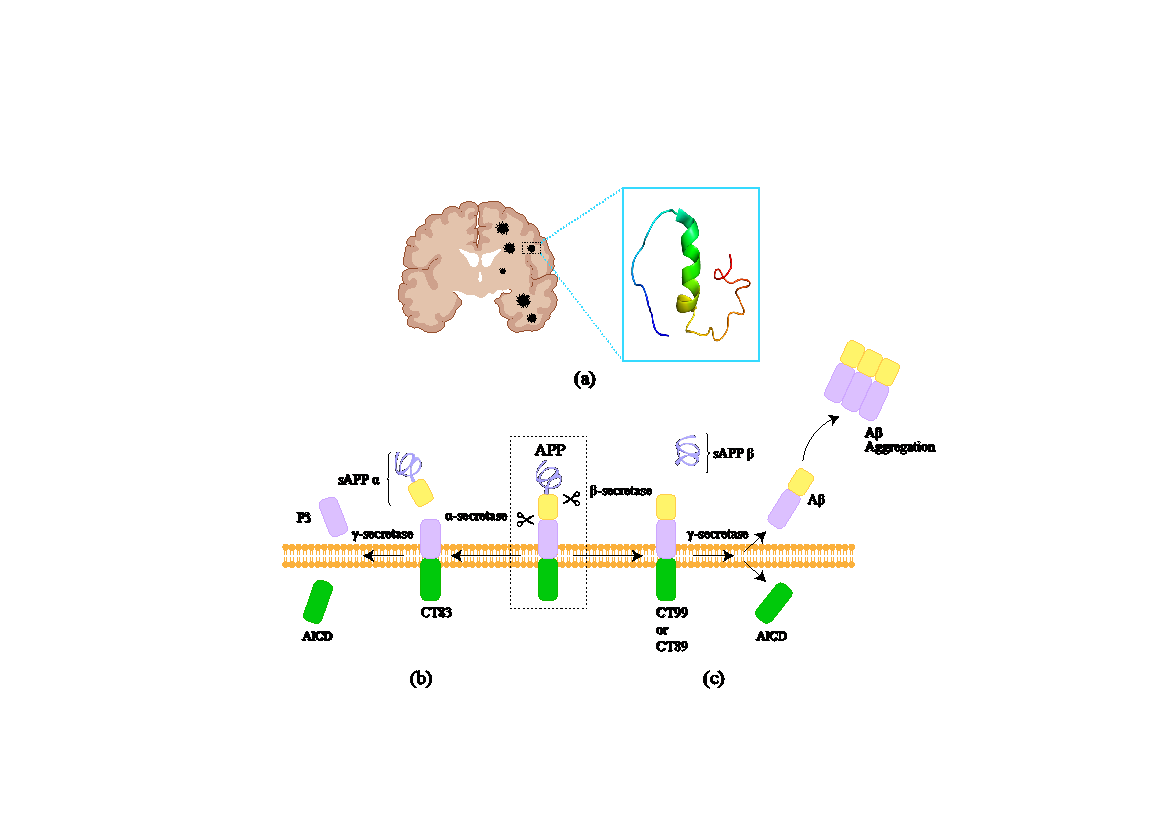
Figure 1. (a) In Alzheimer's patients, altered APP processing results in increased Aβ aggregate accumulation in the brain [23]. (b) In the normal cleavage mechanism of APP on the membrane, the non-amyloidogenic pathway involves APP being first cleaved by α-secretase, producing sAPPα and C83 fragments. These C83 fragments are then cleaved by γ-secretase, yielding non-toxic P3 and AICD. (c) In the abnormal APP processing on the membrane, the amyloidogenic pathway involves APP being first cleaved by β-secretase, producing sAPPβ and C99 fragments. The C99 fragments are then cleaved by γ-secretase, generating toxic Aβ peptides. These peptides aggregate to form fibers in the brain, leading to amyloid plaques, neuronal dsfunction, and cell death.
Aβ is believed to trigger familial Alzheimer's disease and drive its progression [19]. Though initially considered a downstream event of Aβ deposition and tau protein pathologies, tau and Aβ are now thought to act in parallel pathways, potentiating each other's toxic effects and causing Alzheimer's disease [1,20]. Like prion proteins, toxic abnormal conformations of Aβ are generated during the disease and can induce pathological conformations of normal peptides, propagating the disease in the brain [24]. Aβ may also exist in forms such as oligomers or fibrils, with varying toxic effects requiring further study [25,26]. Low-molecular-weight Aβ oligomers seem to interact with various membrane receptors, including prions [26]. However, the significance of these interactions to disease progression remains unclear. Given Aβ's role in Alzheimer's disease, blocking its production via Aβ-degrading enzymes has become a reasonable treatment approach [27,28]. Current research focuses on developing BACE1 and γ-secretase inhibitors, though both have encountered adverse effects in clinical trials [29]. Future studies may improve Aβ reduction effectiveness by modulating γ-secretase to generate shorter, less toxic Aβ versions and combining BACE1 inhibitors [2,28].
2.2. Tau Phosphorylation and Tangle
Tau protein is essential for maintaining microtubule structure and function [10,30]. In Alzheimer's disease (AD), abnormal phosphorylation of Tau causes its dysfunction and the formation of neurofibrillary tangles [31], primarily composed of abnormal Tau proteins [28]. Increased likelihood of Tau protein dissociation from microtubules due to abnormal phosphorylation destabilizes the microtubules and causes them to collapse [31]. Dissociated Tau proteins form neurofibrillary tangles in brain regions associated with memory and cognition, like the hippocampus and cortex [32,33]. While amyloid plaques and neurofibrillary tangles are major features of Alzheimer's disease (AD), their specific mechanisms and relationship are still being studied [28,32]. Tau alterations can independently lead to neurodegenerative changes without Aβ accumulation [34,35]. Initially, Tau alterations were thought to be downstream of Aβ accumulation [36], but now views suggest they may act in parallel pathological pathways leading to AD onset, strengthening their toxic effects [20,37]. Like prions, Tau's toxic conformation arises during AD, inducing pathological conformational changes in normal proteins, allowing disease spread in the brain [1,38,39]. Research and therapeutic strategies targeting Tau are crucial. Potential areas include preventing abnormal Tau phosphorylation, preventing Tau aggregation into tangles, and eliminating already formed Tau tangles using immunotherapy or other methods to mitigate neuronal damage [2,28].
3. Mechanisms of ER Stress in AD
3.1. ER stress response
Endoplasmic reticulum (ER) stress arises from the buildup of unfolded or misfolded proteins in the ER [40,41]. The ER is vital for protein synthesis, folding, modification and transport [42]. ER stress triggers the unfolded protein response (UPR) to restore normal function [43]. If stress persists, the UPR may fail and the cell may initiate apoptosis to protect the organism [14,17].
Recent studies indicate that ER stress is a key factor in several neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD) [14,44]. It's believed to be triggered by pathological proteins like Aβ and tau proteins and further exacerbates their mis-folding and accumulation, forming a vicious cycle [15,45].
3.2. ER stress response and Alzheimer's disease
Amyloid-β aggregation is common in Alzheimer's disease (AD) patients [38], and protein misfolding is the primary cause of endoplasmic reticulum (ER) stress [13]. The relationship between AD and ER stress is complex. ER stress is a cellular stress response caused by misfolded proteins and triggers the unfolded protein response (UPR) to restore homeostasis [41,46]. If ER stress persists, the adaptive mechanism fails, leading to neurological damage [15].
In AD, Aβ production, aggregation, and oligomerization are important factors in disease development. Studies show Aβ oligomers have high neurotoxicity and can induce ER stress [1,19]. ER stress can induce neuroinflammation through UPR activation. This involves release of various factors and chemokines, triggering microglia and astrocyte activation, exacerbating neurodegeneration [14-16].
In Alzheimer's disease (AD), all three major UPR signaling pathways (ATF6, IRE1, and PERK) are activated. Phosphorylated PERK, eIF2α, and IRE1α have been found in AD brains, indicating UPR's significant role in AD pathology [47,48]. Continuous UPR activation triggers cell apoptosis, exacerbating neuronal death and dysfunction [14]. ER stress is also linked to abnormal tau protein phosphorylation and neurofibrillary tangles formation in AD [20]. It promotes tau phosphorylation via the JNK pathway, leading to neuronal dysfunction and death (Figure 2) [48].
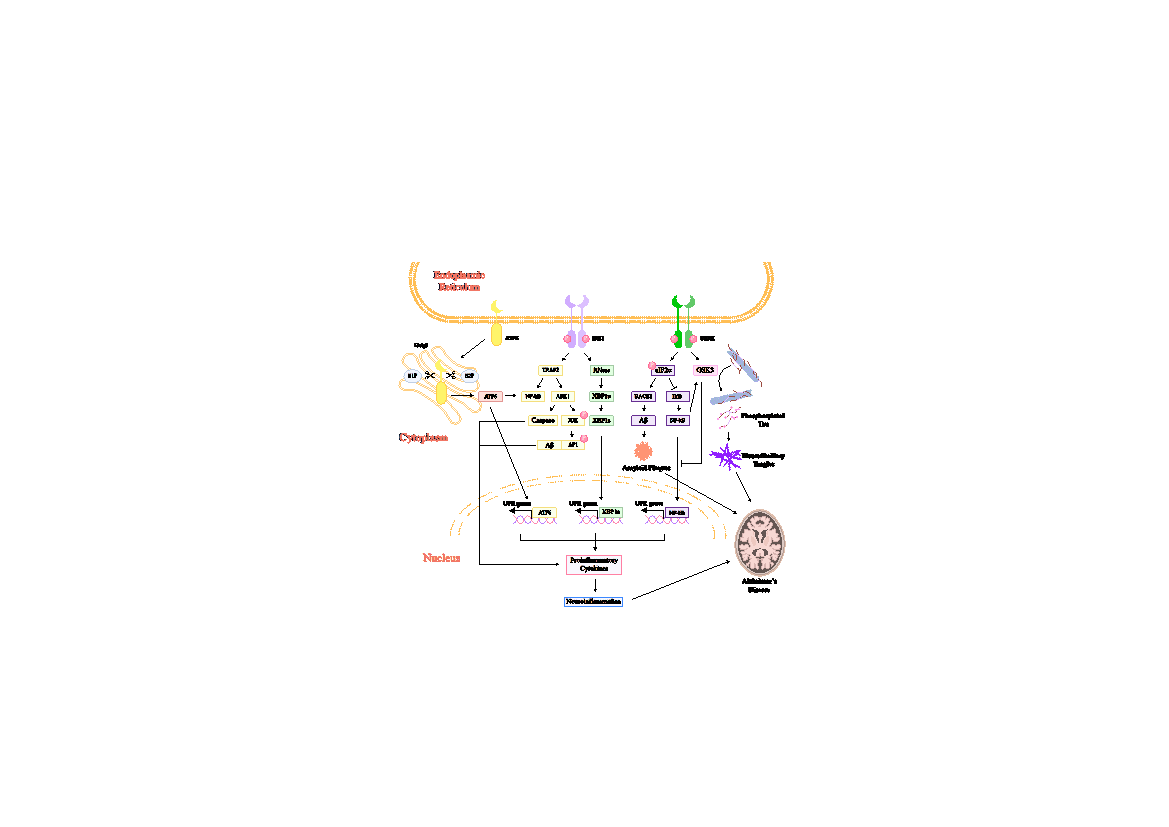
Figure 2. The ER and UPR signalling cascade in neuroinflammation and Alzheimer's disease (AD). The figure illustrates activation pathways of PERK, IRE1, and ATF6, key unfolded protein response (UPR) mediators. PERK activation phosphorylates eIF2α, triggering GSK-3 to form neurofibrillary tangles and phosphorylating BACE1 to generate amyloid-beta (Aβ). eIF2α phosphorylation also activates NF-κB, a pivotal inflammatory transcription factor. IRE1 activation splices XBP1s, induces caspase cascades, and further activates NF-κB, promoting inflammation. ATF6 activation releases its cytosolic domain, translocating to the nucleus and inducing expression of genes driving neuroinflammation and contributing to AD pathology. The diagram highlights these pathways' intersection in driving cellular events underlying AD, including amyloid plaques, neurofibrillary tangles, and chronic inflammation.
3.3. ER stress response and neuroinflammation
The interplay between ER stress and neuroinflammation is highly significant. ER stress triggers adaptive changes, including induction of inflammatory responses [47,50]. The three major signal pathways of the UPR - PERK, IRE1, and ATF6 - play crucial roles in this process. PERK activation leads to eIF2α phosphorylation, subsequently activating NF-κB, a key inflammatory factor. IRE1 activation promotes expression of inflammatory factors through XBP1s translocation and caspase cascade reactions. ATF6 activation releases its cytoplasmic domain, triggering NF-κB and regulating expression of multiple genes implicated in neuroinflammation. These inflammatory responses have a dual role in onset and progression of Alzheimer's disease (AD), potentially protecting neurons and exacerbating neurodegenerative changes [2,51].
3.4. ER stress response and mitochondrial dysfunction
ER stress is closely tied to mitochondrial dysfunction. It can disrupt normal calcium transport from the ER to the mitochondria, causing overloading and malfunction [52]. For example, ER stress can indu12/24/2024 4:54:00 PMce an increase in calcium influx, leading to mitochondrial depolarization and the activation of caspase-12, ultimately triggering cell apoptosis [17,52]. Moreover, ER stress can further impair mitochondrial function by inducing oxidative stress, particularly evident in brains of AD patients [53,54].
3.5. ER stress response and autophagy disorder
The ubiquitin-proteasome pathway is influenced by the ER stress response, affecting AD progression [55]. Autophagy is important for cells to eliminate damaged or misfolded proteins. However, autophagy is often inhibited in AD [57]. Endoplasmic reticulum stress affects autophagy through various pathways, including activation of the UPR and changes in ER-mitochondria contact sites [55]. The IRE1 and PERK pathways in the UPR regulate autophagy gene expression and autophagosome formation. Autophagy dysfunction results in the buildup of misfolded proteins like Aβ and tau, worsening neurodegenerative damage [20,55,57].
The ER stress plays a critical role in the pathogenesis of AD [48], directly causing protein misfolding and neuronal damage, and exacerbating the disease progression through neuroinflammation, mitochondrial dysfunction, and autophagy dysfunction. Understanding the molecular mechanisms of endoplasmic reticulum stress response can elucidate the pathological process of AD, providing important insights for developing novel therapeutic strategies.
3.6. Viral hypothesis of AD and ER stress response
Recent evidence suggests viral infections play a significant role in AD pathogenesis. They trigger neuroinflammation and accelerate AD-related pathological changes through the endoplasmic reticulum stress pathway [5,58]. Some neurotropic viruses, like Zika virus (ZIKV), Japanese encephalitis virus (JEV), herpes simplex virus (HSV), human immunodeficiency virus (HIV), and cytomegalovirus (CMV), can infect and damage neurons in the central nervous system, potentially leading to AD [58-62].
ZIKV infection has gained considerable attention, capable of inducing AD-like pathological features, including Aβ and phosphorylated p-Tau expression, within brain organoid models. ZIKV proteins primarily reside within the endoplasmic reticulum (ER), and replication occurs within the host ER, leading to ER stress and UPR activation. Persistent ER stress from ZIKV infection activates the PERK-eIF2α pathway, ultimately leading to Aβ generation and Tau phosphorylation, key AD pathological features [61,63].
Specifically, sustained activation of the PERK pathway following ZIKV infection leads to phosphorylation of eIF2α, reducing overall protein synthesis while promoting generation of Aβ by increasing abundance of BACE1. Furthermore, GSK3α/β are key in Tau phosphorylation and significantly increase in ZIKV-infected brain organoids, exacerbating Tau pathological changes [64,65]. Use of PERK inhibitors can significantly reduce pathological changes caused by Aβ and p-Tau induced by ZIKV infection, suggesting the PERK-eIF2α pathway is important in ZIKV-induced AD pathology [58,61,63-66].
The Zika virus and some other viral infections accelerate the development of Alzheimer's disease (AD) by inducing endoplasmic reticulum (ER) stress and the unfolded protein response. The discovery of this link not only identifies novel factors in AD pathogenesis but also offers insights into developing new treatment strategies that target the ER stress pathway.
4. Conclusion
Alzheimer's disease is a complex, fatal neurodegenerative disorder marked by Aβ plaque formation and abnormal tau protein phosphorylation, leading to neurofibrillary tangles [1]. These pathological features directly impair neuronal function and survival, and worsen disease progression by inducing endoplasmic reticulum stress and unfolded protein response [6,44,48]. The ER stress not only directly causes protein misfolding and neuronal damage, but also triggers neuroinflammation, mitochondrial dysfunction, and autophagy dysfunction, forming a vicious cycle that further aggravates disease progression [47].
Additionally, viral infections like Zika accelerate the pathological progression of AD by inducing ER stress and UPR [61,63], providing new perspectives on AD's complex pathogenesis. In-depth research on the molecular mechanisms and specific role of endoplasmic reticulum stress response in AD will elucidate its pathology and provide important theoretical foundations and developing new treatment strategies [6].
Future research should investigate ER stress interactions with pathophysiological mechanisms and regulate the UPR pathway to reduce neurotoxicity [7]. Particularly, understanding how viral infections trigger ER stress and pathological changes in AD could provide breakthroughs in AD prevention and treatment [58]. Multi-angle, multi-level research can unravel specific ER stress mechanisms in AD, aiding comprehensive understanding of AD pathogenesis for new treatment insights.
References
[1]. Scheltens, P. et al. Alzheimer’s disease.
[2]. Twarowski, B. & Herbet, M. Inflammatory Processes in Alzheimer’s Disease—Pathomechanism, Diagnosis and Treatment: A Review. International Journal of Molecular Sciences 6518 (2023) doi:10.3390/ijms24076518.
[3]. Roussel, M., Möller, J. & Graeber, M. The case described by Alois Alzheimer in 1911.
[4]. Sosa-Ortiz, A. L., Acosta-Castillo, I. & Prince, M. J. Epidemiology of Dementias and Alzheimer’s Disease. Archives of Medical Research 43, 600–608 (2012).
[5]. 2024 Alzheimer’s disease facts and figures.
[6]. Passeri, E. et al. Alzheimer’s Disease: Treatment Strategies and Their Limitations. International Journal of Molecular Sciences 13954 (2022) doi:10.3390/ijms232213954.
[7]. Elmaleh, DavidR. et al. Developing Effective Alzheimer’s Disease Therapies: Clinical Experience and Future Directions. Journal of Alzheimer’s Disease (2019).
[8]. Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C. & Janke, C. Microtubule-Associated Proteins: Structuring the Cytoskeleton. Trends in Cell Biology 29, 804–819 (2019).
[9]. Martin, L. et al. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Research Reviews 289–309 (2013) doi:10.1016/j.arr.2012.06.003.
[10]. González, A., Singh, S. K., Churruca, M. & Maccioni, R. B. Alzheimer’s Disease and Tau Self-Assembly: In the Search of the Missing Link. International Journal of Molecular Sciences 23, 4192 (2022).
[11]. Neuronal Cell Death. in Encyclopedia of Molecular Pharmacology 1103–1103 (2021). doi:10.1007/978-3-030-57401-7_300365.
[12]. Li, J.-Q., Yu, J.-T., Jiang, T. & Tan, L. Endoplasmic Reticulum Dysfunction in Alzheimer’s Disease. Molecular Neurobiology 51, 383–395 (2015).
[13]. Katayama, T. et al. Induction of neuronal death by ER stress in Alzheimer’s disease. Journal of Chemical Neuroanatomy 28, 67–78 (2004).
[14]. Ghemrawi, R. & Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. International Journal of Molecular Sciences 21, 6127 (2020).
[15]. Hetz, C. & Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nature Reviews Neurology 477–491 (2017) doi:10.1038/nrneurol.2017.99.
[16]. Hetz, C., Zhang, K. & Kaufman, R. J. Mechanisms, regulation and functions of the unfolded protein response. Nature Reviews Molecular Cell Biology 421–438 (2020) doi:10.1038/s41580-020-0250-z.
[17]. Szegezdi, E., Logue, S. E., Gorman, A. M. & Samali, A. Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO reports 7, 880–885 (2006).
[18]. A Self-defeating Anabolic Program Leads to-Cell Apoptosis in Endoplasmic Reticulum Stress-induced Diabetes via Regulation of Amino Acid Flux.pdf.
[19]. O’Brien, R. J. & Wong, P. C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annual Review of Neuroscience 34, 185–204 (2011).
[20]. Zhang, H. et al. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. International Journal of Biological Sciences 2181–2192 (2021) doi:10.7150/ijbs.57078.
[21]. Lanoiselée, H.-M. et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLOS Medicine e1002270 (2017) doi:10.1371/journal.pmed.1002270.
[22]. Zhang, Y., Thompson, R., Zhang, H. & Xu, H. APP processing in Alzheimer’s disease. Molecular Brain 3 (2011) doi:10.1186/1756-6606-4-3.
[23]. Vivekanandan, S., Brender, J. R., Lee, S. Y. & Ramamoorthy, A. A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochemical and Biophysical Research Communications 411, 312–316 (2011).
[24]. Jucker, M. & Walker, L. C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nature Neuroscience 1341–1349 (2018) doi:10.1038/s41593-018-0238-6.
[25]. Lee, S. J. C., Nam, E., Lee, H. J., Savelieff, M. G. & Lim, M. H. Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors. Chemical Society Reviews 310–323 (2016) doi:10.1039/c6cs00731g.
[26]. Amyloid toxicity in Alzheimer’s disease.
[27]. Leissring, M. A. Aβ-Degrading Proteases: Therapeutic Potential in Alzheimer Disease. CNS Drugs 30, 667–675 (2016).
[28]. Hong-Qi, Y., Zhi-Kun, S. & Sheng-Di, C. Current advances in the treatment of Alzheimer’s disease: focused on considerations targeting Aβ and tau. Translational Neurodegeneration 1, (2012).
[29]. Hampel, H. et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biological Psychiatry 745–756 (2021) doi:10.1016/j.biopsych.2020.02.001.
[30]. Barbier, P. et al. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Frontiers in Aging Neuroscience (2019) doi:10.3389/fnagi.2019.00204.
[31]. Wang, J.-Z., Xia, Y.-Y., Grundke-Iqbal, I. & Iqbal, K. Abnormal Hyperphosphorylation of Tau: Sites, Regulation, and Molecular Mechanism of Neurofibrillary Degeneration. Journal of Alzheimer’s Disease 33, S123–S139 (2012).
[32]. Furcila, D., DeFelipe, J. & Alonso-Nanclares, L. A Study of Amyloid-β and Phosphotau in Plaques and Neurons in the Hippocampus of Alzheimer’s Disease Patients. Journal of Alzheimer’s Disease 64, 417–435 (2018).
[33]. Furcila, D., Domínguez-Álvaro, M., DeFelipe, J. & Alonso-Nanclares, L. Subregional Density of Neurons, Neurofibrillary Tangles and Amyloid Plaques in the Hippocampus of Patients With Alzheimer’s Disease. Frontiers in Neuroanatomy 13, (2019).
[34]. Mechanisms of tau-induced neurodegeneration.
[35]. Baki, L. et al. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. The EMBO Journal 23, 2586–2596 (2004).
[36]. Hardy, J. & Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends in Pharmacological Sciences 383–388 (1991) doi:10.1016/0165-6147(91)90609-v.
[37]. Li, K. et al. Synaptic Dysfunction in Alzheimer’s Disease: Aβ, Tau, and Epigenetic Alterations. Molecular Neurobiology 55, 3021–3032 (2018).
[38]. Ashrafian, H., Zadeh, E. H. & Khan, R. H. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. International Journal of Biological Macromolecules 382–394 (2021) doi:10.1016/j.ijbiomac.2020.11.192.
[39]. Penke, B., Szűcs, M. & Bogár, F. Oligomerization and Conformational Change Turn Monomeric β-Amyloid and Tau Proteins Toxic: Their Role in Alzheimer’s Pathogenesis. Molecules 25, 1659 (2020).
[40]. Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum.
[41]. Boyce, M. & Yuan, J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death & Differentiation 363–373 (2006) doi:10.1038/sj.cdd.4401817.
[42]. The Role of the Endoplasmic Reticulum in Protein Synthesis, Modification and Intracellular Transport.
[43]. Hetz C., Zhang K. & Kaufman R. Mechanisms, regulation and functions of the unfolded protein response.
[44]. Lindholm, D., Wootz, H. & Korhonen, L. ER stress and neurodegenerative diseases. Cell Death & Differentiation 13, 385–392 (2006).
[45]. Uddin, Md. S., Yu, W. S. & Lim, L. W. Exploring ER stress response in cellular aging and neuroinflammation in Alzheimer’s disease. Ageing Research Reviews 70, 101417 (2021).
[46]. Kaufman, R. J., Back, S. H., Song, B., Han, J. & Hassler, J. The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation inβ‐cells. Diabetes, Obesity and Metabolism 12, 99–107 (2010).
[47]. Uddin, Md. S. et al. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Molecular Neurobiology 57, 2902–2919 (2020).
[48]. Ajoolabady, A., Lindholm, D., Ren, J. & Pratico, D. ER stress and UPR in Alzheimer’s disease: mechanisms, pathogenesis, treatments. Cell Death & Disease (2022) doi:10.1038/s41419-022-05153-5.
[49]. JNK: A Stress-Activated Protein Kinase Therapeutic Strategies and Involvement in Alzheimer’s and Various Neurodegenerative Abnormalities.
[50]. From endoplasmic-reticulum stress to the inflammatory response.
[51]. Inflammation in Neurodegenerative Disease—A Double-Edged Sword.
[52]. Takuma, K., Yan, S. S., Stern, D. M. & Yamada, K. Mitochondrial Dysfunction, Endoplasmic Reticulum Stress, and Apoptosis in Alzheimer’s Disease. Journal of Pharmacological Sciences 97, 312–316 (2005).
[53]. Alqahtani, T. et al. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and Amyotrophic Lateral Sclerosis -An updated review. Mitochondrion 71, 83–92 (2023).
[54]. Bhandary, B., Marahatta, A., Kim, H.-R. & Chae, H.-J. An Involvement of Oxidative Stress in Endoplasmic Reticulum Stress and Its Associated Diseases.
[55]. Cai, Y. et al. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 12, 225–244 (2016).
[56]. Autophagy: cellular and molecular mechanisms.
[57]. Krishnan, S. et al. Activate or Inhibit? Implications of Autophagy Modulation as a Therapeutic Strategy for Alzheimer’s Disease. International Journal of Molecular Sciences 21, 6739 (2020).
[58]. Bruno, F. et al. Alzheimer’s disease as a viral disease: Revisiting the infectious hypothesis.
[59]. Honjo, K., van Reekum, R. & Verhoeff, N. P. L. G. Alzheimer’s disease and infection: Do infectious agents contribute to progression of Alzheimer’s disease? Alzheimer’s & Dementia 5, 348–360 (2009).
[60]. A preliminary neuropathological study of Japanese encephalitis in humans and a mouse model.
[61]. Zika Virus Infection of Human Mesenchymal Stem Cells Promotes Differential Expression of Proteins Linked to Several Neurological Diseases.
[62]. Neurocognition and the Aging Brain in People With HIV: Implications for Screening.
[63]. Lee, S.-E. et al. Zika virus infection accelerates Alzheimer’s disease phenotypes in brain organoids. Cell Death Discovery 8, (2022).
[64]. N-Methyl- D-Aspartate (NMDA) Receptor Blockade Prevents Neuronal Death Induced by Zika Virus Infection.
[65]. Amyloid precursor protein is a restriction factor that protects against Zika virus infection in mammalian brains.
[66]. Zika Virus.
Cite this article
Zhou,Y. (2025). The Role of Endoplasmic Reticulum Stress in Alzheimer's Disease: Molecular Mechanisms and Viral Hypotheses. Theoretical and Natural Science,78,1-9.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Scheltens, P. et al. Alzheimer’s disease.
[2]. Twarowski, B. & Herbet, M. Inflammatory Processes in Alzheimer’s Disease—Pathomechanism, Diagnosis and Treatment: A Review. International Journal of Molecular Sciences 6518 (2023) doi:10.3390/ijms24076518.
[3]. Roussel, M., Möller, J. & Graeber, M. The case described by Alois Alzheimer in 1911.
[4]. Sosa-Ortiz, A. L., Acosta-Castillo, I. & Prince, M. J. Epidemiology of Dementias and Alzheimer’s Disease. Archives of Medical Research 43, 600–608 (2012).
[5]. 2024 Alzheimer’s disease facts and figures.
[6]. Passeri, E. et al. Alzheimer’s Disease: Treatment Strategies and Their Limitations. International Journal of Molecular Sciences 13954 (2022) doi:10.3390/ijms232213954.
[7]. Elmaleh, DavidR. et al. Developing Effective Alzheimer’s Disease Therapies: Clinical Experience and Future Directions. Journal of Alzheimer’s Disease (2019).
[8]. Bodakuntla, S., Jijumon, A. S., Villablanca, C., Gonzalez-Billault, C. & Janke, C. Microtubule-Associated Proteins: Structuring the Cytoskeleton. Trends in Cell Biology 29, 804–819 (2019).
[9]. Martin, L. et al. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Research Reviews 289–309 (2013) doi:10.1016/j.arr.2012.06.003.
[10]. González, A., Singh, S. K., Churruca, M. & Maccioni, R. B. Alzheimer’s Disease and Tau Self-Assembly: In the Search of the Missing Link. International Journal of Molecular Sciences 23, 4192 (2022).
[11]. Neuronal Cell Death. in Encyclopedia of Molecular Pharmacology 1103–1103 (2021). doi:10.1007/978-3-030-57401-7_300365.
[12]. Li, J.-Q., Yu, J.-T., Jiang, T. & Tan, L. Endoplasmic Reticulum Dysfunction in Alzheimer’s Disease. Molecular Neurobiology 51, 383–395 (2015).
[13]. Katayama, T. et al. Induction of neuronal death by ER stress in Alzheimer’s disease. Journal of Chemical Neuroanatomy 28, 67–78 (2004).
[14]. Ghemrawi, R. & Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. International Journal of Molecular Sciences 21, 6127 (2020).
[15]. Hetz, C. & Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nature Reviews Neurology 477–491 (2017) doi:10.1038/nrneurol.2017.99.
[16]. Hetz, C., Zhang, K. & Kaufman, R. J. Mechanisms, regulation and functions of the unfolded protein response. Nature Reviews Molecular Cell Biology 421–438 (2020) doi:10.1038/s41580-020-0250-z.
[17]. Szegezdi, E., Logue, S. E., Gorman, A. M. & Samali, A. Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO reports 7, 880–885 (2006).
[18]. A Self-defeating Anabolic Program Leads to-Cell Apoptosis in Endoplasmic Reticulum Stress-induced Diabetes via Regulation of Amino Acid Flux.pdf.
[19]. O’Brien, R. J. & Wong, P. C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annual Review of Neuroscience 34, 185–204 (2011).
[20]. Zhang, H. et al. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. International Journal of Biological Sciences 2181–2192 (2021) doi:10.7150/ijbs.57078.
[21]. Lanoiselée, H.-M. et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLOS Medicine e1002270 (2017) doi:10.1371/journal.pmed.1002270.
[22]. Zhang, Y., Thompson, R., Zhang, H. & Xu, H. APP processing in Alzheimer’s disease. Molecular Brain 3 (2011) doi:10.1186/1756-6606-4-3.
[23]. Vivekanandan, S., Brender, J. R., Lee, S. Y. & Ramamoorthy, A. A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochemical and Biophysical Research Communications 411, 312–316 (2011).
[24]. Jucker, M. & Walker, L. C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nature Neuroscience 1341–1349 (2018) doi:10.1038/s41593-018-0238-6.
[25]. Lee, S. J. C., Nam, E., Lee, H. J., Savelieff, M. G. & Lim, M. H. Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors. Chemical Society Reviews 310–323 (2016) doi:10.1039/c6cs00731g.
[26]. Amyloid toxicity in Alzheimer’s disease.
[27]. Leissring, M. A. Aβ-Degrading Proteases: Therapeutic Potential in Alzheimer Disease. CNS Drugs 30, 667–675 (2016).
[28]. Hong-Qi, Y., Zhi-Kun, S. & Sheng-Di, C. Current advances in the treatment of Alzheimer’s disease: focused on considerations targeting Aβ and tau. Translational Neurodegeneration 1, (2012).
[29]. Hampel, H. et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biological Psychiatry 745–756 (2021) doi:10.1016/j.biopsych.2020.02.001.
[30]. Barbier, P. et al. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Frontiers in Aging Neuroscience (2019) doi:10.3389/fnagi.2019.00204.
[31]. Wang, J.-Z., Xia, Y.-Y., Grundke-Iqbal, I. & Iqbal, K. Abnormal Hyperphosphorylation of Tau: Sites, Regulation, and Molecular Mechanism of Neurofibrillary Degeneration. Journal of Alzheimer’s Disease 33, S123–S139 (2012).
[32]. Furcila, D., DeFelipe, J. & Alonso-Nanclares, L. A Study of Amyloid-β and Phosphotau in Plaques and Neurons in the Hippocampus of Alzheimer’s Disease Patients. Journal of Alzheimer’s Disease 64, 417–435 (2018).
[33]. Furcila, D., Domínguez-Álvaro, M., DeFelipe, J. & Alonso-Nanclares, L. Subregional Density of Neurons, Neurofibrillary Tangles and Amyloid Plaques in the Hippocampus of Patients With Alzheimer’s Disease. Frontiers in Neuroanatomy 13, (2019).
[34]. Mechanisms of tau-induced neurodegeneration.
[35]. Baki, L. et al. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. The EMBO Journal 23, 2586–2596 (2004).
[36]. Hardy, J. & Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends in Pharmacological Sciences 383–388 (1991) doi:10.1016/0165-6147(91)90609-v.
[37]. Li, K. et al. Synaptic Dysfunction in Alzheimer’s Disease: Aβ, Tau, and Epigenetic Alterations. Molecular Neurobiology 55, 3021–3032 (2018).
[38]. Ashrafian, H., Zadeh, E. H. & Khan, R. H. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. International Journal of Biological Macromolecules 382–394 (2021) doi:10.1016/j.ijbiomac.2020.11.192.
[39]. Penke, B., Szűcs, M. & Bogár, F. Oligomerization and Conformational Change Turn Monomeric β-Amyloid and Tau Proteins Toxic: Their Role in Alzheimer’s Pathogenesis. Molecules 25, 1659 (2020).
[40]. Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum.
[41]. Boyce, M. & Yuan, J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death & Differentiation 363–373 (2006) doi:10.1038/sj.cdd.4401817.
[42]. The Role of the Endoplasmic Reticulum in Protein Synthesis, Modification and Intracellular Transport.
[43]. Hetz C., Zhang K. & Kaufman R. Mechanisms, regulation and functions of the unfolded protein response.
[44]. Lindholm, D., Wootz, H. & Korhonen, L. ER stress and neurodegenerative diseases. Cell Death & Differentiation 13, 385–392 (2006).
[45]. Uddin, Md. S., Yu, W. S. & Lim, L. W. Exploring ER stress response in cellular aging and neuroinflammation in Alzheimer’s disease. Ageing Research Reviews 70, 101417 (2021).
[46]. Kaufman, R. J., Back, S. H., Song, B., Han, J. & Hassler, J. The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation inβ‐cells. Diabetes, Obesity and Metabolism 12, 99–107 (2010).
[47]. Uddin, Md. S. et al. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Molecular Neurobiology 57, 2902–2919 (2020).
[48]. Ajoolabady, A., Lindholm, D., Ren, J. & Pratico, D. ER stress and UPR in Alzheimer’s disease: mechanisms, pathogenesis, treatments. Cell Death & Disease (2022) doi:10.1038/s41419-022-05153-5.
[49]. JNK: A Stress-Activated Protein Kinase Therapeutic Strategies and Involvement in Alzheimer’s and Various Neurodegenerative Abnormalities.
[50]. From endoplasmic-reticulum stress to the inflammatory response.
[51]. Inflammation in Neurodegenerative Disease—A Double-Edged Sword.
[52]. Takuma, K., Yan, S. S., Stern, D. M. & Yamada, K. Mitochondrial Dysfunction, Endoplasmic Reticulum Stress, and Apoptosis in Alzheimer’s Disease. Journal of Pharmacological Sciences 97, 312–316 (2005).
[53]. Alqahtani, T. et al. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and Amyotrophic Lateral Sclerosis -An updated review. Mitochondrion 71, 83–92 (2023).
[54]. Bhandary, B., Marahatta, A., Kim, H.-R. & Chae, H.-J. An Involvement of Oxidative Stress in Endoplasmic Reticulum Stress and Its Associated Diseases.
[55]. Cai, Y. et al. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 12, 225–244 (2016).
[56]. Autophagy: cellular and molecular mechanisms.
[57]. Krishnan, S. et al. Activate or Inhibit? Implications of Autophagy Modulation as a Therapeutic Strategy for Alzheimer’s Disease. International Journal of Molecular Sciences 21, 6739 (2020).
[58]. Bruno, F. et al. Alzheimer’s disease as a viral disease: Revisiting the infectious hypothesis.
[59]. Honjo, K., van Reekum, R. & Verhoeff, N. P. L. G. Alzheimer’s disease and infection: Do infectious agents contribute to progression of Alzheimer’s disease? Alzheimer’s & Dementia 5, 348–360 (2009).
[60]. A preliminary neuropathological study of Japanese encephalitis in humans and a mouse model.
[61]. Zika Virus Infection of Human Mesenchymal Stem Cells Promotes Differential Expression of Proteins Linked to Several Neurological Diseases.
[62]. Neurocognition and the Aging Brain in People With HIV: Implications for Screening.
[63]. Lee, S.-E. et al. Zika virus infection accelerates Alzheimer’s disease phenotypes in brain organoids. Cell Death Discovery 8, (2022).
[64]. N-Methyl- D-Aspartate (NMDA) Receptor Blockade Prevents Neuronal Death Induced by Zika Virus Infection.
[65]. Amyloid precursor protein is a restriction factor that protects against Zika virus infection in mammalian brains.
[66]. Zika Virus.