







1. Introduction
Accounting for more than half of the people’s total protein in blood, human serum albumin(HSA) is one of the richest proteins in human body, which plays an indispensable role in maintaining blood osmolality balance, material transport, antioxidant and other physiological processes. Decrease in HSA levels may cause a series of liver and kidney diseases, such as cirrhosis, nephritis, etc. Apart from that, cardiovascular diseases like heart failure also have a connection with the reduction. Therefore, an in-depth look at the structural features and physiological functions of HSAs not only helps us form a better understanding of its role in health and disease at the molecular level, but also possesses a significant meaning in revealing the pathogenesis of related diseases, guiding clinical treatment strategies, and promoting drug development. Additionally, the versatility of HSA offers bright future for its application in biopharmaceuticals, drug delivery systems, etc. Through a deeper understanding of their structure and function, more efficient drug carriers can be designed to improve the efficacy and safety of drugs. Meanwhile, the potential applications of HSA in disease treatment, such as plasma replacement therapy and cellular repair, provide new ideas and methods for clinical treatment.
And for that reason, many scholars conducted investigations on human serum albumin, including the nature and structure of the protein and drug development under different mechanisms previously. However, drug design or related mechanistic studies based on existing targets are based on traditional experimental methods, including, but not limited to, the following five: (1) HSA nanoparticles prepared by fine tuning of pH, salt concentration and cross-linking agent have excellent stability and high drug transport efficiency; Ru-ATRA-HSANP nanoparticles can be effectively taken up by cells and have significant anti-tumor metastasis effects[1] (2) Elevated concentrations of the complexes linearly burst HSA fluorescence, and the bursting effect decreases with increasing temperature, suggesting that hydrophobic and hydrogen bonding dominate the binding; these complexes also significantly alter the α-helical structure of HSA, providing a potential structural target for antitumor drug development[2] (3) The stability of covalent adducts formed by drugs with HSA is affected by pH, nucleophilicity and steric factors, acting through mechanisms such as Michael addition. Covalent tyrosine kinase inhibitors (TKIs) interact with target proteins through a two-step binding mode, and their plasma stability correlates with adverse drug reactions, providing new perspectives for drug safety assessment[3] (4) Human serum albumin (HSA) is a non-glycosylated heart-shaped α-helical protein with long-term blood presence and the ability to transport a wide range of substances. The helical structure of HSA is stabilized upon ligand binding, potentially generating an induced circular dichroism signal. Its hydrophobic interactions, particularly the hydrophobic environment of Trp-214 in substructural domain II A, are essential for ligand binding[4] (5) HSA, a key transport protein in human plasma, is a disulfide bond-rich peptide that activates muscle satellite cells to promote muscle growth and repair. The reducing environment of Escherichia coli is not conducive to the proper formation and stability of HSA disulfide bonds, whereas the eukaryotic host system successfully expresses soluble rHSA through slow translation and oxidative folding at low temperatures[5].
However, there are limitations in the above methods: firstly, all the studies are case by case treatment of drugs rather than systematic analysis, and the research ideas and experimental design methods of single studies do not have universal value; secondly, the traditional biological experimental methods have the limitations of long time-consuming experiments and high costs, and the experimental conclusions derived from the experiments are prone to systematic errors, which may lead to a decrease in the accuracy of the final results. In order to overcome the above shortcomings, this paper adopts a new approach of artificial intelligence in order to systematically and completely analyze the protein structure prediction, and then discover new insights to better assist the drug design and development of the target.
2. Methodology
2.1. Model Introduction
This paper focuses on the structure prediction of human serum albumin with the help of alphafold2, which is a deep learning model based on computer big data that outputs a 3D spatial structure with atomic accuracy by inputting the amino acid sequence of a protein[6]. The model consists of four parts, namely MSA module, Evoformer, structure module and Recycling, in which the MSA module extracts the sequence features between amino acids and amino acid pairs, and compares them with the homologous proteins in the original gene database to get the amino acid sequence features to be studied; Evoformer acts as the decoding module to predict the structure of proteins. The Evoformer acts as a decoder to edit the features obtained from the MSA module into the shape that the neural network wants to express. At this time, although the size of the input and output of the model has not changed significantly, the tensor and dimension of the two three-dimensional matrices can characterise the dependency relationship between each amino acid and the weighting matrix very well; the decoder structure module is responsible for transforming the Evoformer inputs into the structure module. The decoder structure module is responsible for transforming the Evoformer inputs into 3D results that can be visualised, which is achieved by deriving the corresponding atomic coordinates of each atom, and at the same time the structure module outputs the confidence of the model results to evaluate its own results, with different values of confidence corresponding to different colours, and the differences in colours are shown in the final visualised 3D structure. The final Recycling will turn the output of Evoformer and structure module into the input of Evoformer to guarantee the accuracy of the output result at the atom, and usually the model will be traversed three times again to improve the prediction accuracy and confidence of the model. It is worth noting that although the model overall achieves four cycles for the main workflow, it does not add too much running burden to the computer, and it is one of the promising tools for protein model structure prediction and future development in the field of pharmaceuticals and medicine.
Existing research suggests that Alphafold2's work is of cross-generational significance. Designing AlphaFold2 using a combination of bioinformatics and physical techniques allows us to construct components using physical and geometric inductive biases to learn from PDB data and minimise hand-crafted features, and high-precision models such as this one greatly advance the development of scientists' research on protein functional analysis, expanding the range of downstream applications of proteins for future The development of antibiotics, targeted drugs, cancer, viral infections and enzyme research and development will make pioneering contributions to future research.
2.2. Workflow
As a protein crucial in maintaining blood osmotic pressure and transporting substances, human albumin is widely found in human blood and has important physiological functions in immune regulation, hormone delivery, and acid-base balance in the body, etc. Therefore, the aim of this study is to start from the intact structure of the protein of HSA and explore whether there are more novel structural features or potentially applicable ligands to bind with HSA to form new drugs for the benefit of mankind.
Firstly, the sequence information of the standard HSA protein named P02768-ALBU_HUMAN was retrieved from the protein database uniprot, and the amino acid sequence feature of length 609 was obtained, and then with the help of the bioinformatics tool alphafold2, the obtained complete sequence information was input into the online prediction platform to predict the complete full-length protein 3D structure with atomic precision. The Alphafold2 model outputs the 3D stereospiral protein structure while scoring the predicted local distance difference test scores with reference to the structural confidence level, so as to judge the credibility of the prediction results. After visualisation of the HSA protein structure, the work invoked the Plip tool for structural analysis, which is capable of returning a list of interactions detected at the single atom level without any structural pre-processing, including seven types of hydrogen bonds, hydrophobic contacts, π-stacking, π-cation interactions, salt bridges, water bridges and halogen bonds[7]. plip is used as a rule-based plip was used as a rule-based detection tool to personalise the analysis of the HSA protein structure. Then, based on the secondary structure of HSA proteins and the specific rotation angle, plip will investigate whether they will form hydrophobic bonds or bind to the pockets and ligands of the corresponding drug molecules at specific positions, and then conduct downstream analyses to search for the possible targets of HSA proteins, and then confirm the structural angle again, which will ultimately form a closed-loop process to find the promising drugs for the development of the cause of human health.
3. Results
By inputting the standard HSA protein sequence information called P02768-ALBU_HUMAN with the length of 609 retrieved from the protein database into the alphafold2 model and comparing it with the big database, we get the conclusions as shown in the following pictures. The overall curve of Figure 1 is higher, except for the amino acid sequences with a slight decrease in the head and the tail, and the rest of the amino acid matches to the homologous protein information is around 900 on average, indicating that the MSA module of alphafold2 has richer coevolutionary information from the original database during the system operation, and the human serum albumin has higher sequence coverage in the alphafold2 database, and the prediction results are more accurate.
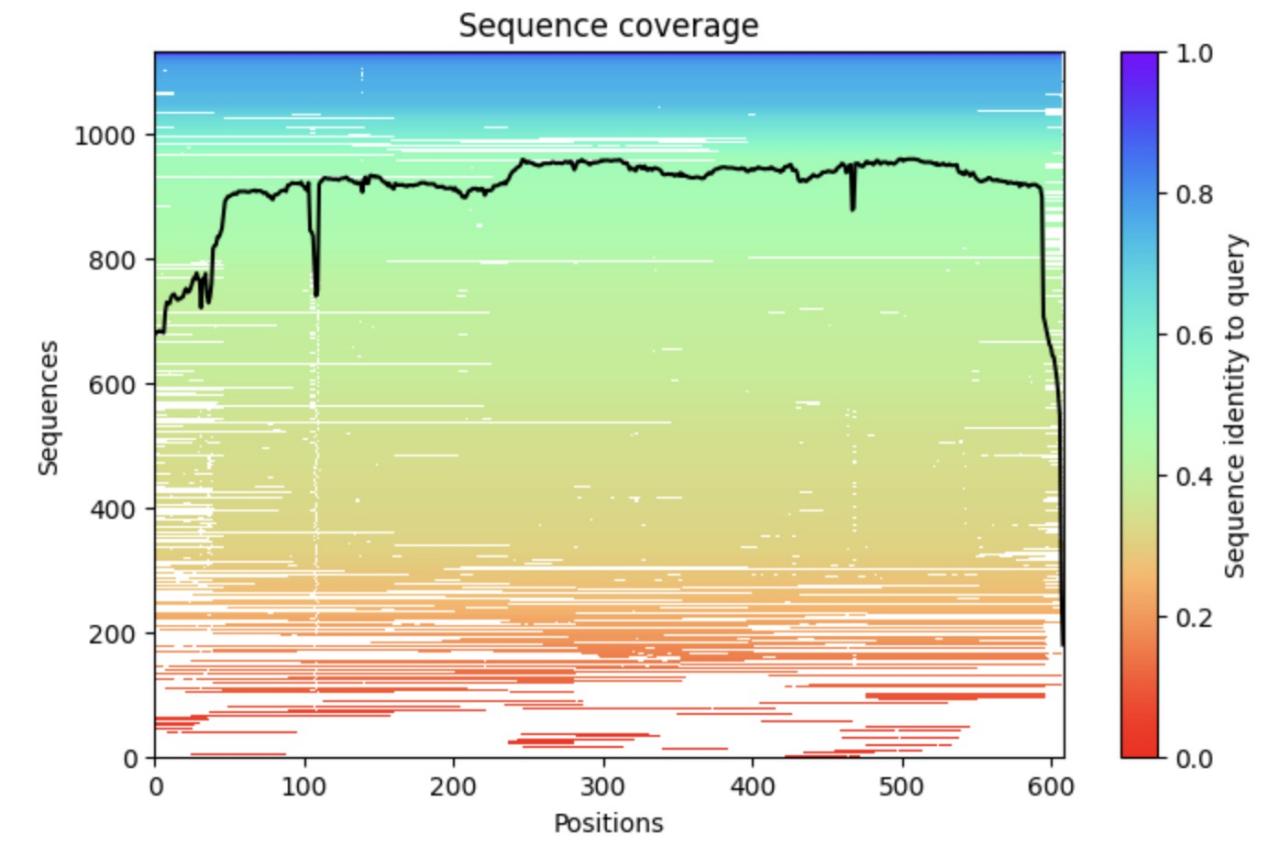
Figure 1. The sequence coverage of the HSA in alphafold2 database.
a, The horizontal axis indicates each position on the sequence, and the vertical axis indicates the homologous protein information that can be found in the original database. FIGURE overall indicates that amino acids at most positions starting from the front end of the protein can be matched to more sequences in the database, the HSA protein is richer in homologous protein information, and the MSA module finds more coevolutionary information.
Figure 2 presents the prediction results of the alphafold2 model in 3D visualisation In the 3D visualisation of the predicted structure of HSA, it is found that almost all the proteins show curly shapes, in which the simultaneous appearance of alpha-helix and beta-sheet fully indicates that the predicted structure of HSA possesses rich secondary structures and typical structural domains different from the monolinear form; the ingenious pockets formed downstream of the proteins on the lower right part of the picture also greatly increase the possibility of the binding of HSA itself as an active research component with other drugs or ligands, and it is a protein with multiple structural domains. It is a very promising target with multiple binding sites. At the same time, the coiled folding between proteins of the same species is also an outward manifestation of hydrogen bonding or other non-covalent bonding interactions between the various components, which can be demonstrated by the plip tool at a later stage.
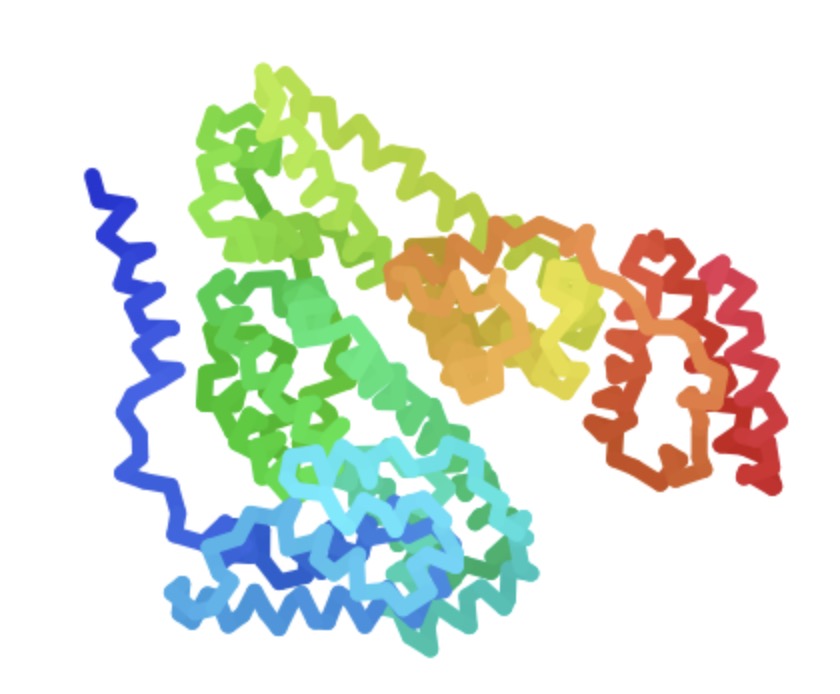
Figure 2. Structure of human serum proteins obtained by alphafold2 prediction.
a, The predicted structure of HSA shows that the protein has a large number of alpha-helix secondary structures greater than ten at the head and tail ends and in the full length of the sequence, in addition to the middle domain part of the protein still has a triple-folded beta-sheet conformation while maintaining multiple alpha-helix coils; the downstream of the protein has a clear notch formed by the coiled folding of the protein. The formation of a distinct groove in the downstream fold of the protein is a reflection of hydrogen bonding or other non-covalent bonding interactions between the various parts of the HSA protein.
Alphafold2 calls five different and well-trained models randomly initialised behind the server web-side model prediction, and different PLDDT results are obtained from multiple runs due to the random information seed settings during system runtime, based on the five models' scores, which are named rank 1, rank 2, rank 3 and rank 4 in order from top to bottom, rank 5, represented by different colours in figure 3. Although the performance of the five models is almost the same, the actual structural analysis or downstream prediction validation preferred to refer to the highest confidence score of the rank 1 model. the overall trend distribution of Figure 3 is normal and high confidence, which can be fully trusted and subsequent analysis.
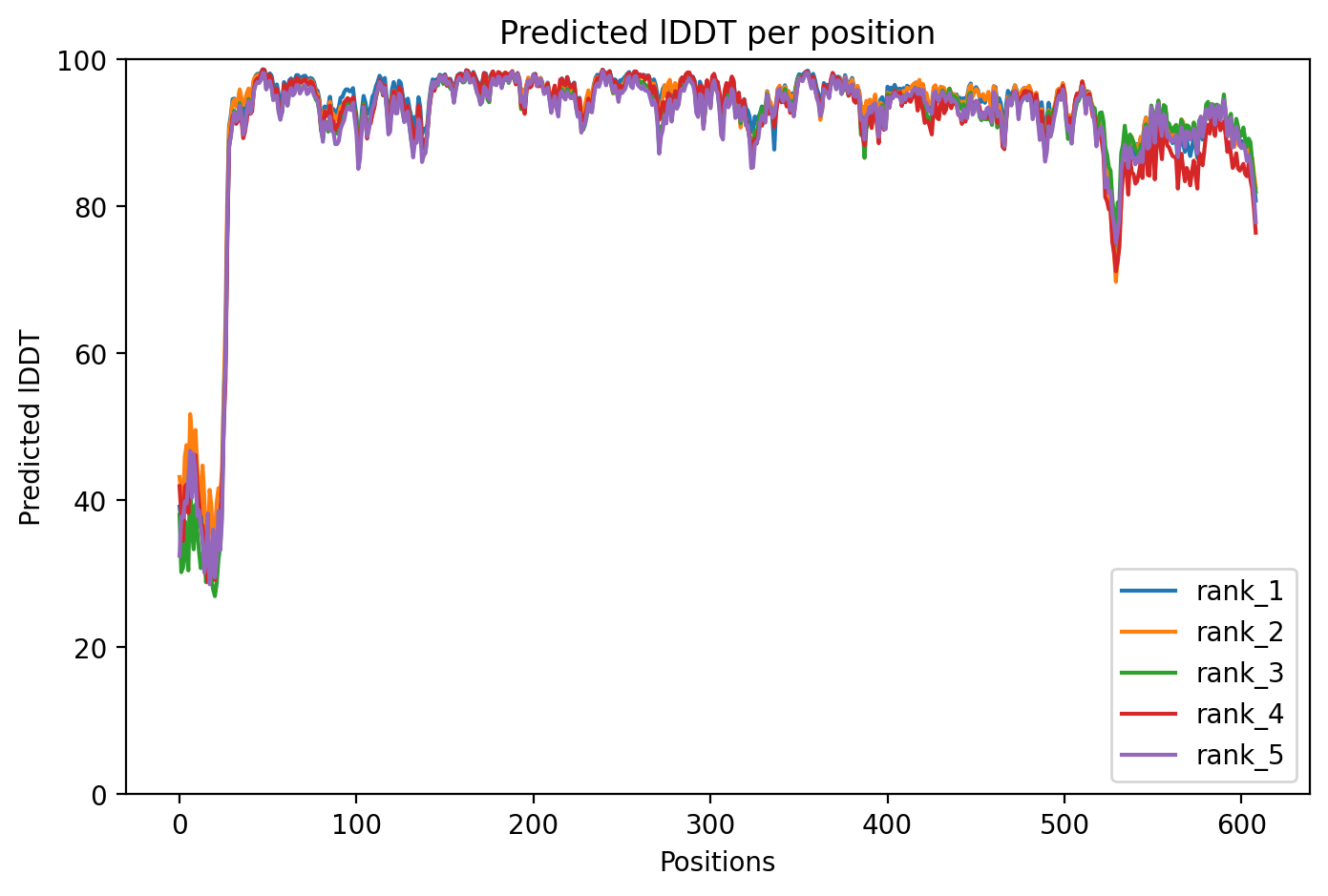
Figure 3. Predicted IDDT per position of human serum proteins obtained by alphafold2 prediction.
a, The horizontal axis represents each position 0-609 on the amino acid sequence of the HSA protein, and the vertical axis represents the PLDDT scores that reflect the confidence of the model predictions, and the rank 1 to rank 5 icons in the lower right corner represent the five independent models with high to low confidence, with the values ranging from 0 to 100. The results of the prediction images show that the PLDDT scores of the overall five models are high, except for the PLDDT score of the anterior end, which is located at 40-60 due to the high flexibility of the protein head and tail ends, the scores of the vertical coordinates of the horizontal coordinates from 50 to 520 are around 90, and even the confidence scores of some local segments are close to 100, which is a highly-confidence prediction, indicating the high accuracy of structural prediction at the location. structure is predicted with high accuracy.
Figure 4 presents content that mirrors that of Figure 3, both analysing from the perspective of position which positions on the protein have better confidence in the structural prediction of the result, with a higher degree of confidence. Overall the best module has a PLDDT score of 91 and a pTM score of 0.865, which is a good prediction, and it is precisely because the amino acid subfragments of the HSA proteins correspond to significantly strong co-evolutionary information that their sequence dataset can be predicted very quickly with an obvious local structure.

Figure 4. Predicted average error per position of human serum proteins obtained by alphafold2 prediction.
a, The horizontal and vertical axes represent the amino acids in each position, which is a pairwise concept, and the coordinates range from 0-609, the bluer the colour of the image, the lower the error of the model prediction result, and the redder it is, the higher it is. 0-30 is a large position with a large error, the colour is red, the prediction result is relatively inaccurate, and the result of the error is large; the diagonal line symbolises the error of the position and its own morphological characteristics, and the dark blue colour indicates that the error is zero. The diagonal line symbolises the error at the position and its own morphological characteristics, dark blue means the error is 0.
The predicted uptake and distribution structure data of the interaction of the human albumin named 4E99 with additional small molecules was called into the plip tool to analyse which amino acids at key positions achieve binding to the drug molecule through which specific non-covalent bonding form. Figure 5 serves as a small illustration of the application of the plip tool to show the potential binding sites when the interaction with the drug named P8S-A-601 occurs during the bonding process. illustration, showing the potential binding sites of the drug named P8S-A-601 when it interacts with the HSA protein during the bonding process. Based on structural prediction, although some proteins are structurally continuous, the amino acid sequence is not necessarily continuous, and this figure also serves as a figurative demonstration of this phenomenon, showing the discontinuous state of the potential binding amino acids on the HSA protein. Thus, the many amino acid ‘breakpoints’ found at the observed positions in figure 5 are essentially due to their proximity to the original predicted structure.
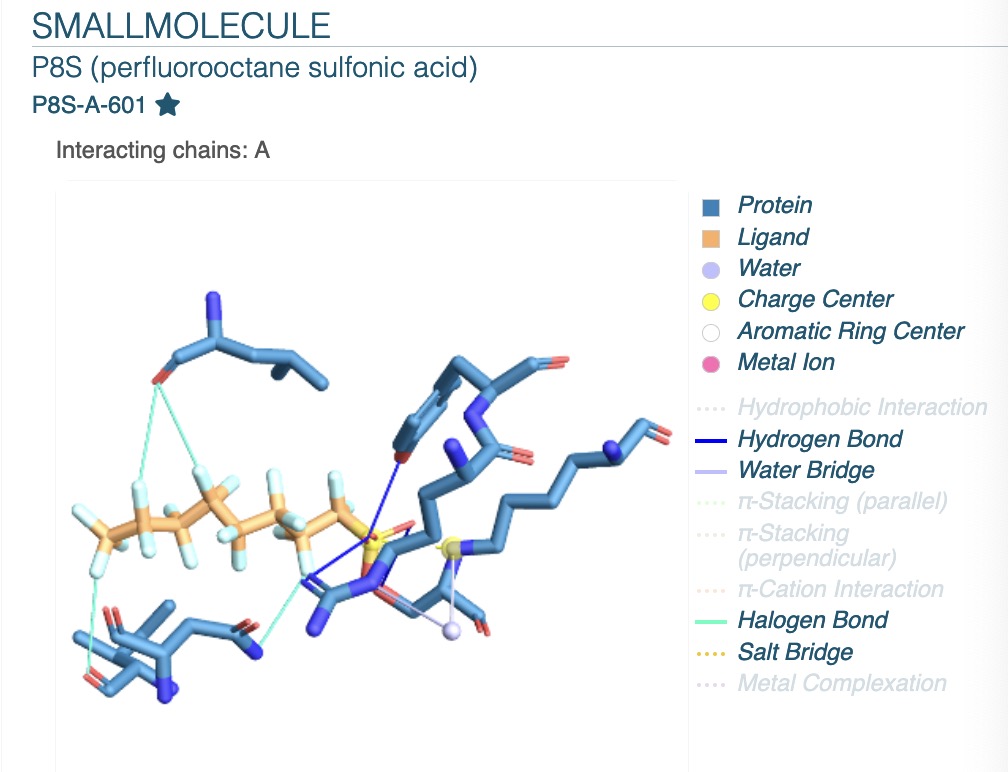
Figure 5. The types of non-covalent bonding interactions of amino acids obtained after plip calls.
a, The plip tool is known to predict several non-covalent bond types shown in the bottom right corner of the image, where the colour void indicates that the type of action form is not detected in the protein structure, while the legend bottom colour shows the actual colour indicating that the non-covalent bond type can be detected at the sequence position of the specific amino acid. In the image chains A represents the protein HSA. Based on the legend, the blue region is human albumin and the orange colour is its ligand small molecule, the focus of this study is mainly on the structure of the blue protein itself.
The following table presents the specific data from the results of the plip run, with Residues representing the specific amino acid sequence positions at which the interactions occur, AA being the abbreviation for amino acids, and Interaction being the specific type of non-covalent bonding interaction. Each row of data in the table can be illustrated with the sentence ‘AA amino acids at the residues position of human albumin readily interact with drug molecules in the form of interactions’, e.g., the amino acid Arginine at position 410 is more likely to form hydrogen bonds with its corresponding ligand drug. For example, the Arginine amino acid at position 410 is more likely to form hydrogen bonds with its corresponding ligand drug, so for drug molecules or proteins that are prone to form hydrogen bonds, Arginine at position 410, Tyrosine at position 411, and serine at position 489 are the key binding sites, and Halogen Bonds are the places where halogen bonds are formed with ligands at position 388, Halogen bonds are formed with the ligand at positions 388, 391, 430 and 324, which correspond to the four signature amino acids of Isoleucine, Asparagine, Leucine and asparatic acid. Except for ARG at position 209, which can additionally form salt bridges, the LYS amino acid at position 414 can form both water bridges and salt bridges with drug ligands.
Table1. Specific types of action and sequence positions of amino acid binding sites derived from plip.
Index | Residue | AA | Interaction |
1 | 410A | ARG | Hydrogen Bonds |
2 | 411A | TYR | Hydrogen Bonds |
3 | 489A | SER | Hydrogen Bonds |
4 | 414A | LYS | Water Bridges |
5 | 388A | ILE | Halogen Bonds |
6 | 391A | ASN | Halogen Bonds |
7 | 430A | LEU | Halogen Bonds |
8 | 414A | LYS | Salt Bridges |
9 | 324A | ASP | Halogen Bonds |
10 | 209A | ARG | Salt Bridges |
The redisue indicates the specific sequence position at which the amino acids of human albumin can interact with the drug, the AA represents the type of amino acid at that location, and the interaction describes the type of interaction that occurs on binding.
The vast majority of protein-to-protein binding or protein-drug interactions are based on the physical formation of non-covalent bonds, with hydrogen and hydrophobic bonds accounting for a large proportion. No atomic exchange is involved between the two, and the low energy generated by the protein and the drug molecule as a whole during binding makes the drug adsorbed stably, while the inconsistency between the local spatial structure and the intrinsic properties such as polarity, charge and hydrophobicity of its side chain itself leads to the influence of the corresponding side-chain groups and physicochemical properties, and thus the formation of non-covalent bonding is not the same type of action.
The specific positions in the table not only represent only the high probability of binding to the drug-ligand molecule, but also indicate that the contiguous amino acid regions in the vicinity of the position can be recognised as having HIGHLY-POSSIBLE potential binding sites. Compared with the results of the alphafold2 prediction presented in Figure 1, the sequence coverage of amino acids in the part of the sequence coding 380-430 is higher, and the scores of the corresponding models of PLDDT are higher, although the specific scores fluctuate slightly up and down, but they are all above 80, indicating that the credibility of the overall macromodel prediction of the structure at this location is higher, and that the prediction results based on the plip tool can be considered as a potential binding site for drug ligands. The prediction based on plip tool yielded more accurate local pocket sites that are easy to bind.
This will be very advantageous for future downstream workers in matching suitable drug molecules for drug design based on geometrical structures. Workers can focus on the local binding epitopes where the above key sites are located, use sampling and energy optimisation methods for molecular docking, put all the known molecules in the drug library or newly designed drugs into the local sites, and match the shapes and physicochemical properties, etc., with a view to finding the most suitable pocket of drugs.
4. Conclusion
In summary, starting from the related diseases that may be caused by insufficient HSA in the human body, this paper has achieved the 3D structure prediction of human serum albumin with atomic accuracy by using the HSA information sequences in the uniprot protein database and the AI tool of alphafold2, and then combined with the output results, we analysed the potential binding sites of this protein through the conclusions related to the non-covalent bonding information of ligand binding returned by the plip tool. The conclusions related to the interaction information returned by the plip tool were used to analyse the potential binding sites of the protein. These binding sites will be used as key hotspots that will be crucial in the downstream stage when going for structure-based design of drug backbones and linking the hotspots together, providing an important reference for the formation of potential ligands for drug studies that are structurally matched to the interbonding of HSA.
This application of combining protein biological structure prediction with AI artificial intelligence technology can solve many problems that cannot be broken through in biomedical experiments, greatly reducing complex and repetitive physical experiments, improving the efficiency of drug research and development, accelerating the process of biological analysis, which is very favourable to the design of the future repositioning of drugs, and providing an innovative and cutting-edge idea reference for the exploration of the side-effects in the later development of the drugs. It is a vivid example of interdisciplinary integration in the new era and a direct response to new health challenges, which is beneficial to the development of human health.
References
[1]. Ding, X., Shi, H. D. & Liu, Y. C.. (2021). Construction and anti-tumour metastatic effects of Ru(III) and all-trans retinoic acid co-transported nanodrugs using human serum albumin as a carrier. Journal of Higher Education Chemistry (10), 3040-3046.
[2]. Yang Jing, Li Li, Liang Jiandan, Huang Shan, Su Wei, Wei Yashu... & Xiaoqi. (2023). Study on the interaction mechanism of aminothiourea aryl ruthenium complexes with human serum albumin. Spectroscopy and Spectral Analysis (09), 2761-2767.
[3]. Liu, Xiaoyun, Diao, Xingxing & Zhong, Dafang. (2024). Advances in the identification of covalent adducts of drugs with human serum albumin. Journal of Pharmacy (04), 886-898. doi:10.16438/j.0513-4870.2023-0922.
[4]. Tripathi, M., Chauhan, S., Princess, R., Khan, R. H., Siddiqi, M. K., Syed, R., Kalam, M. A., Guha, S., & Sarkar, A. (2024). Binding interaction of four azo linked copper (II) complexes with Human Serum Albumin (HSA): Spectroscopic and molecular docking explorations. Results in Chemistry, 9, 101637. https://doi.org/10.1016/j.rechem.2024.101637
[5]. Cho, Y. J., Kim, H., & Lim, S. I. (2024). Preserved Structure and Function of Human Serum Albumin Self-folded in the Oxidative Cytoplasm of Escherichia coli. Journal of Biotechnology, 390, 62–70. https://doi.org/10.1016/j.jbiotec.2024.05.005
[6]. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. a. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., . . . Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
[7]. Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F., & Schroeder, M. (2015). PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Research, 43(W1), W443–W447. https://doi.org/10.1093/nar/gkv315
Cite this article
Wang,Y. (2025). Facilitating structure-based drug design for human albumin by Artificial Intelligence. Theoretical and Natural Science,78,67-75.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Ding, X., Shi, H. D. & Liu, Y. C.. (2021). Construction and anti-tumour metastatic effects of Ru(III) and all-trans retinoic acid co-transported nanodrugs using human serum albumin as a carrier. Journal of Higher Education Chemistry (10), 3040-3046.
[2]. Yang Jing, Li Li, Liang Jiandan, Huang Shan, Su Wei, Wei Yashu... & Xiaoqi. (2023). Study on the interaction mechanism of aminothiourea aryl ruthenium complexes with human serum albumin. Spectroscopy and Spectral Analysis (09), 2761-2767.
[3]. Liu, Xiaoyun, Diao, Xingxing & Zhong, Dafang. (2024). Advances in the identification of covalent adducts of drugs with human serum albumin. Journal of Pharmacy (04), 886-898. doi:10.16438/j.0513-4870.2023-0922.
[4]. Tripathi, M., Chauhan, S., Princess, R., Khan, R. H., Siddiqi, M. K., Syed, R., Kalam, M. A., Guha, S., & Sarkar, A. (2024). Binding interaction of four azo linked copper (II) complexes with Human Serum Albumin (HSA): Spectroscopic and molecular docking explorations. Results in Chemistry, 9, 101637. https://doi.org/10.1016/j.rechem.2024.101637
[5]. Cho, Y. J., Kim, H., & Lim, S. I. (2024). Preserved Structure and Function of Human Serum Albumin Self-folded in the Oxidative Cytoplasm of Escherichia coli. Journal of Biotechnology, 390, 62–70. https://doi.org/10.1016/j.jbiotec.2024.05.005
[6]. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. a. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., . . . Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
[7]. Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F., & Schroeder, M. (2015). PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Research, 43(W1), W443–W447. https://doi.org/10.1093/nar/gkv315