







1. Introduction
The Uvaria genus comprises mostly climbing shrubs distributed across tropical habitats across Africa, Asia, and Australia. This genus, known for its biological activities, is widely implemented in traditional medicine [1]. For instance, Uvaria angolensis, categorised as nontoxic according to OECD guidelines, demonstrates antiplasmodial effects and treats malaria [2]. Uvaria angolensis bark extracts specifically(delete this) also inhibit HIV-1 genome replication [3]. U. cherrevensis roots are used to treat urinary disorders [4]. U. dulcis stem, and U. rufa roots treats fevers and kidney failures [5]. Due to the widespread ethnobotanical implementations and desirable biological activities of the Uvaria genus, ethnopharmacological research into phytochemicals derived from Uvaria leptopoda could lead to new drug discoveries. Uvaria leptopoda (King) J. Sinclair is a climber found in tropical areas of Thailand [6]. Phytochemical investigations into Uvaria leptopoda remain limited, with the discovery of uvarialeptones A-C and Uvarialeptols A & B published only recently in May of 2024. Like other species of the Uvaria genus, these derivatives are all highly oxygenated cyclohexenes with potential medicinal applications [7,8]. Research into Uvaria leptopoda derivatives has already led to findings in ethnopharmacology. Evaluation regarding the cytotoxicity of uvarialeptones A and C against SNU-1 human gastric cancer cells showed IC50 values on par with the clinical drug cisplatin. Due to gastric cancer cells potentially developing cisplatin resistance, implementing uvarialeptones A and C may improve chemotherapy targeting SNU-1 gastric cancer cells. Additionally, uvarialeptone A demonstrated an IC50 value of 18 μM, lower than cisplatin's 25 μM. Uvarialeptone C also demonstrated minimal toxicity and potent anti-inflammatory potential due to the inhibiting RAW264.7 macrophage nitric oxide production. Furthermore, uvarialeptone C’s IC50 value of 6.7 μM is significantly smaller than diclofenac sodium’s IC50 of 331 μM, the current industry standard for anti-inflammatory treatment [8]. The potential ethnopharmacological significance of such phytochemical derivatives prompts research regarding retrosynthetic analysis and synthesis proposals. Successful chemical synthesis of Uvaria leptopoda derivatives could streamline further biological assay and potential preclinical trials, as it could provide an alternative production method more efficient than separation from Uvaria leptopoda extracts. The aim of this paper is to develop an outline for the synthesis of uvarialeptone A and C retrosynthetically and use said outline to propose a viable total synthesis pathway from simple, common, or commercially available precursors.
2. Retrosynthetic Analysis of Uvarialeptone A and C
2.1. Overview of Uvarialeptone A and C
Uvarialeptone C consists of four six-member rings, two of which are aromatic and one of which is oxygenated, like tetrahydropyran. Uvarialeptone C has three fused six-member rings comprising of an aromatic ring, an oxygenated ring, and a cyclohexane. A methoxy group is attached to the aromatic phenyl ring in a para position to the oxygen of the oxygenated ring. Both bridgehead carbons between the oxygenated ring and cyclohexane have a methoxy group attached. The cyclohexane also contains two ketone groups attached in the meta positions relative to the oxygen of the oxygenated ring. The α carbon in the cyclohexane positioned between both carbonyl groups has a three-carbon chain attached and participates in a conjugated diene structure across four carbons. An alcohol group is attached to the alkene closest to the α carbon with E isomerism. A phenyl group is attached to the other end of the three-carbon chain trans to the rest of the molecule. Uvarialeptone C is structurally similar to uvarialeptone A, which essentially comprises of two uvarialeptone C molecules. A reaction catalysed by visible light would allow two benzylic alkenes of two uvarialeptone C molecules to form a cyclobutane, leading to uvarialeptone A [9]. Hence, discussions regarding the synthesis of either compound apply to both in practice.
2.2. Retrosynthesis of Uvarialeptone A and C
As discussed above, the ideal initial disconnection of uvarialeptone A would be of two carbon single bonds of the central cyclobutane. As shown in Figure 1, such a disconnection in compound 1 could result in two identical uvarialeptone C molecules (compound 2). The resulting compound should have a phenyl group trans to the rest of the molecule. Synthesis could achieve this disconnection via a visible light-driven [2+2] cycloaddition. The following disconnection of the carbon side chain attached to the α carbon between two carbonyls would be efficient, as the disconnection is more convergent in nature. This should result in overall higher synthesis yields. As seen in Figure 2, such a disconnection would result in compound 3, a system with three fused rings, and compound 4, which is 3-phenyl propanal. Hence, compound 4 would be a target molecule for the forward synthesis of both uvarialeptones. Such a disconnection could be replicated in synthesis through LDA aldol addition, oxidation, and keto-enol tautomerisation.
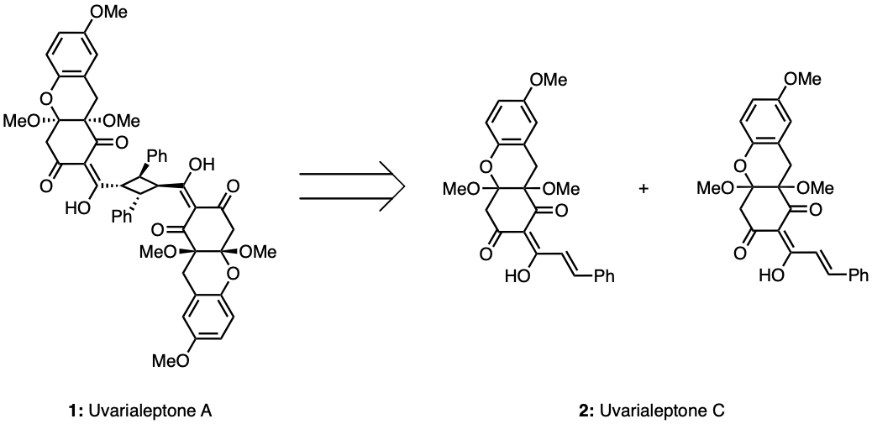
Figure 1. Uvarialeptone A & C.
Afterwards, a functional group conversion of compound 4 from methoxy groups into hydroxy groups would be necessary. These disconnections result in compound 5, as seen in Figure 2. Although the disconnections of ether functional groups are fine-tuning and linear in nature, simultaneously converting all three alcohols is much more efficient than individual conversions. All three disconnections are achievable in forward synthesis via any etherification reaction involving methanol.
Compound 5 should naturally undergo ring chain tautomerisation, resulting in the formation of a ketone and the disconnection of an oxygenated ring, leading to compound 6. In forward synthesis, the ring tautomer should be favoured in equilibrium and could thus provide a considerable yield [10].
Following the tautomerisation, compound 6 could disconnect two α carbon and carbonyl bonds, separating a formaldehyde molecule and resulting in compound 7. This is achievable in synthesis via LDA aldol reaction and oxidation. The tertiary alcohol in compound 7 is attached to two identical carbonyl-containing chains. Hence, there are two 1,2 deoxygenation patterns within the compound. Two oxygens could be disconnected or eliminated from compound 7, leaving an alkene, as seen in compound 8. This could be replicated in synthesis with osmium tetroxide and an oxidation reaction. Compound 8 is clearly comprised of a hydroquinone with a carbon chain attached, as seen in Figure 3. Disconnecting the single bond between the aromatic carbon and the sp3 hybridised carbon would, in this case, maximise efficiency, as such a disconnection constitutes convergent synthesis. The disconnection will separate compound 8 into compounds 9 and 10. Compound 9 is hydroquinone, which is common and commercially available. Compound 10 will be another target molecule for the synthesis of uvarialeptones. Such a disconnection would be available in synthesis via a Friedel-Crafts alkylation reaction, hence why Compound 10 is chlorinated.
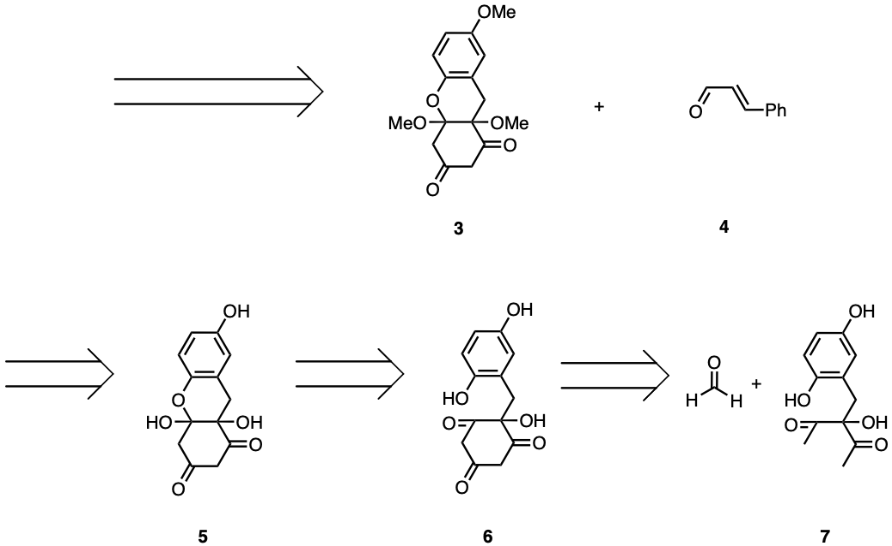
Figure 2. Retrosynthesis of Uvarialeptone A & C.
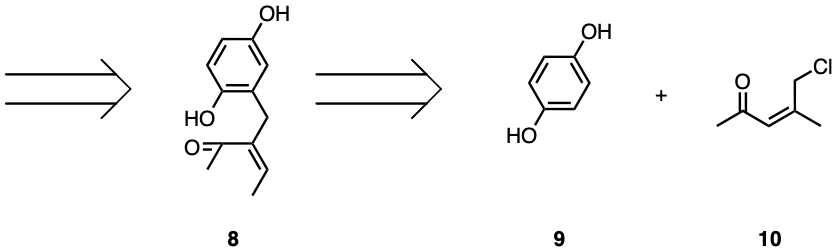
Figure 3. Retrosynthesis Target Molecules.
3. Proposed Synthesis of Uvarialeptone A
3.1. Target molecules
Based on the above retrosynthetic analysis, the actual synthesis would require compounds 4, 9, and 10. The following paper will outline the synthesis of compounds 4 and 10, though it should be noted that compound 4 is commercially available as cinnamaldehyde.
3.2. Synthesis of Fragment: Compound 10
Compound 10 contains a hidden 1,3 di-oxygenation pattern. The pentene carbon backbone of the compound could be synthesised through a LDA-catalysed addition between equal parts acetone and acetaldehyde. The deprotonated α carbon present in acetone should attack the δ+ carbonyl carbon present in acetaldehyde, thus forming a five-carbon chain with a 1,3 di-oxygenation pattern as seen in compound 11.
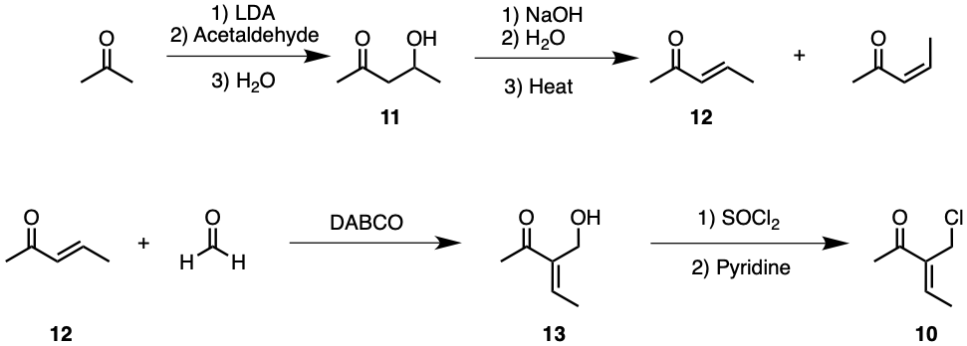
Figure 4. Synthesis of Compound 10.
Admittedly, due to the reactivity of aldehydes, it is possible that acetaldehyde may react with another molecule itself rather than acetone. Acetone may even attack two acetaldehyde molecules, forming a heptane chain instead. However, since acetone contains two α carbons while acetaldehyde contains only one, it is still probable for acetone to attack acetaldehyde. The overall synthesis should be minimally affected even if the yield is not particularly high, because acetone and acetaldehyde are widely available, and this is also the first step of the synthesis.
Following the LDA aldol reaction, compound 11 may undergo an aldol condensation reaction, forming compound 12. Notably, the alkene functional group only forms between α and β carbons because the reaction is catalysed by NaOH, which only deprotonates α carbons. The geometric isomerism of compound 12 is insignificant, as the alkene functional group will eventually be reacted with osmium tetroxide.
Compound 12 may undergo the Baylis-Hillman reaction with formaldehyde and catalyst DABCO to form a side chain, as seen in compound 13 [11]. Despite aldehyde's reactivity, formaldehyde should not be able to react with molecules of itself under given conditions. Hence, the reaction yield should be considerable. The Baylis-Hillman reaction may alter the product's geometric isomerism, which is once again insignificant.
Compound 13 is structurally identical to target molecule compound 10. In the presence of SOCl2 and pyridine, compound 13 may undergo functional group interconversion, forming an alkyl halide. Compound 10 will later undergo Friedel-Crafts alkylation.
3.3. Synthesis of Fragment: Compound 4
Compound 4 contains a trans alkene functional group between an aldehyde and phenyl group, as seen in Figure 5. The phenyl group could be obtained using benzoic acid as a starting material.
Benzoic acid reacts with methanol and acid under heat to undergo Fischer esterification, resulting in compound 14. Compound 14 may then react with methyl ethanoate in the presence of sodium methoxide and methanol to undergo a crossed Claisen condensation, joining both esters together. Compound 14 (Methyl benzoate) is non-enolisable, as its only α carbon participates in aromaticity. Hence, compound 15 should have high yields. Nevertheless, excess methyl benzoate is recommended for maximum yield.
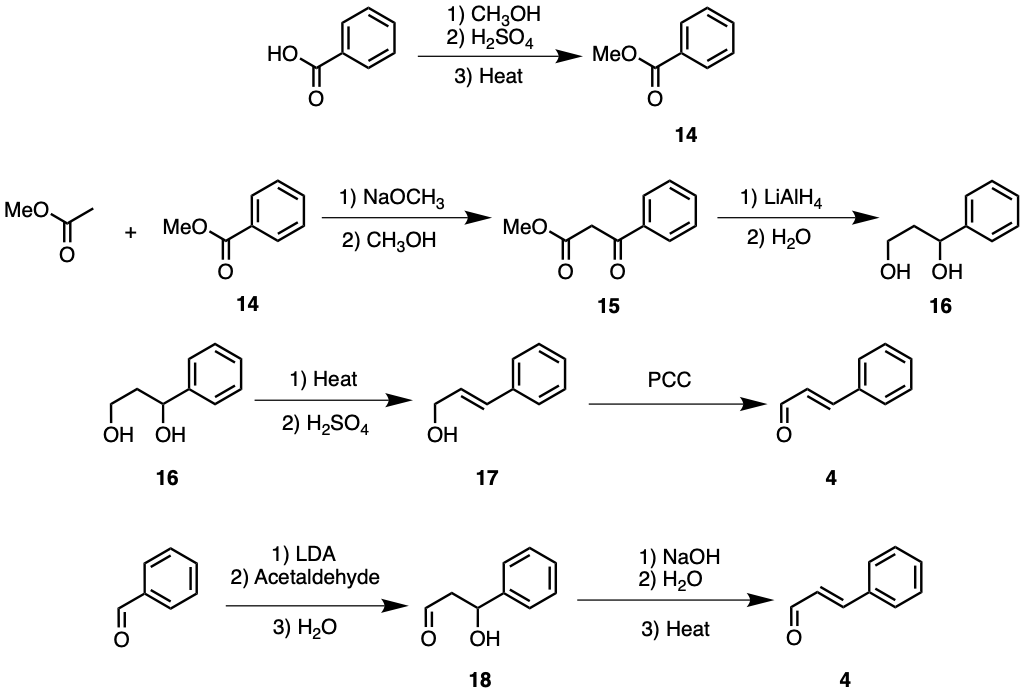
Figure 5. Synthesis of Compound 4.
Compound 15 may react with LiAlH4 and water, reducing both ester and ketone functional groups to alcohols, resulting in compound 16. Compound 16 could be heated with H2SO4 to undergo E1 elimination, forming compound 17. The E1 mechanism rarely occurs at primary carbons; hence, the elimination is almost guaranteed to occur at the secondary alcohol level. However, geometric isomerism poses a problem. The trans-geometric isomer should be thermodynamically favoured, though a significant portion of the yield may be of the cis isomer instead. PCC could oxidise compound 17 to target molecule compound 4.
Alternatively, compound 4 could be synthesised from phenyl methanal as a starting material. Phenyl methanal could undergo LDA-catalysed aldol reaction with acetaldehyde, forming compound 18. Compound 18 may then be treated with NaOH and water and heated to undergo aldol condensation, forming compound 4. Once again, elimination will only occur between α and β carbons. However, the geometric isomerism will further lower yields, similar to the prior synthesis pathway.
The latter synthesis seems more straightforward, but as discussed before, aldehydes are highly reactive and unpredictable. Acetaldehyde could once again react with molecules of itself, lowering yields. The former scheme avoids using aldehydes by using esters and Claisen condensation instead. Nevertheless, an absolute comparison of efficiencies between either scheme is possible only through practical investigations. In the former synthesis pathway, the yield of trans isomers could perhaps be increased by using a larger ester alkyl chain. The methyl ethanoate could be replaced with a cyclohexyl ethanoate. LiAlH4 could be replaced with NaBH4 to create a hemiacetal instead. Since cyclohexyl groups are significantly larger than methyl groups, the trans isomer will be significantly more thermodynamically favoured than the cis isomer due to steric repulsion. The new compound could then be treated with DIBAL and PCC to produce compound 4. It should also be noted that compound 4 is commercially available as cinnamaldehyde.
3.4. Synthesis of Uvarialeptone A
Compound 9, or hydroquinone, is widely commercially available and thus justifies as a starting material. The initial step of the synthesis involving compound 9 would be to attach silyl-protecting groups by adding two equivalents of trimethyl silyl and triethyl amine, resulting in compound 19, as seen in Figure 6. This ensures that the alcohol will not be accidentally oxidised.
Afterwards, compound 19 could react with one equivalent of compound 10 and AlCl3, undergoing Friedel-Crafts alkylation, producing compound 20. Compound 19’s symmetry ensures equivalence of all four carbons available to alkylation. This maximises yields as long as only one equivalent of compound 10 is added to each compound 19.
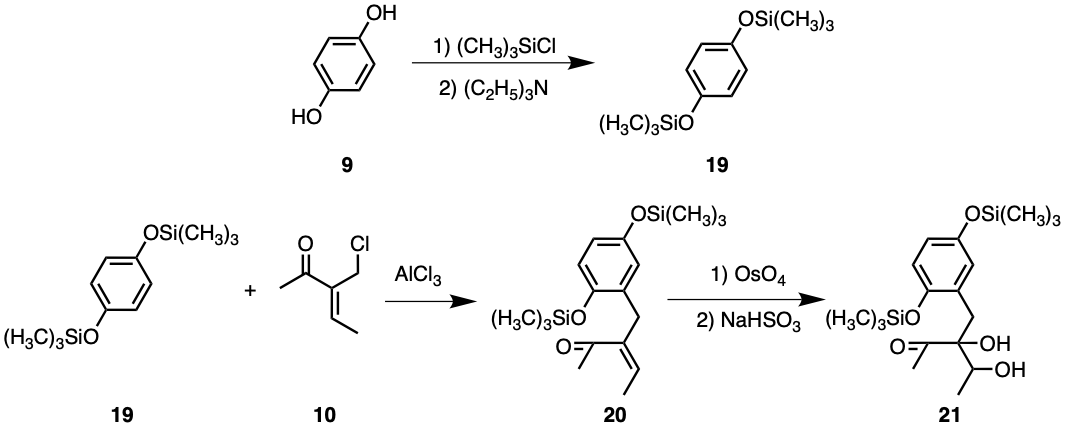
Figure 6. Synthesis of Uvarialeptone A & C Part 1.
Osmium tetroxide may then be added to compound 20 with NaHSO3, forming compound 21. This justifies the insignificance of geometric isomerism present in compound 10, as both sp2 hybridised carbons are converted to sp3. The chirality of the alcohol adjacent to the ketone functional group, however, is significant, but this will be addressed later.
The addition of PCC to compound 21 will oxidise the secondary alcohol into a ketone, as seen in compound 22. Hence, the chirality of the secondary alcohol’s carbon centre in compound 21 was insignificant, as seen in Figure 7.
Compound 22 could be reacted with LDA and acetaldehyde in the presence of water and PCC, completing the cyclohexane seen in uvarialeptones. The LDA-catalysed aldol reaction occurs twice, once intermolecularly and once intramolecularly, forming compound 23. LDA will initially deprotonate a primary α carbon, which then attacks the formaldehyde molecule intermolecularly via an aldol reaction. Formaldehyde’s aldehyde functional group is converted to an alcohol as a result. Hence, PCC is needed to oxidise the alcohol back to an aldehyde. Afterwards, LDA will deprotonate the remaining primary α carbon again, which then attacks the δ+ carbonyl carbon intramolecularly, completing the cyclohexane. An alcohol is formed via an aldol reaction, though it is immediately oxidised to a ketone by PCC.
As the reactions from compound 21 to compound 23 all require PCC as a reagent, it may be possible to combine two reactions into one by adding 2 equivalents of PCC initially. Despite formaldehyde's reactiveness, it should not be able to undergo aldol reactions with molecules of itself, so the impact on yields should be minimal.
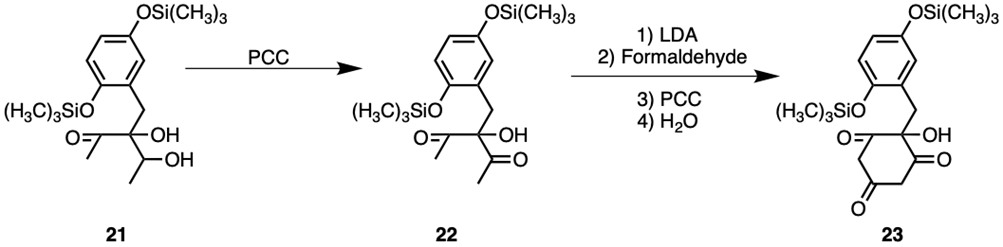
Figure 7. Synthesis of Uvarialeptone A & C Part 2.
After the completion of all oxidation reactions, TBAF may be added to compound 23 to remove the silyl-protecting groups, resulting in compound 6. Compound 6 undergoes ring/chain tautomerisation, forming an equilibrium with compound 5, as seen in Figure 8. However, since ring-form tautomers are significantly more favoured compared to their chain-form counterparts, compound 5 should be the predominant tautomer in equilibrium [12]. The chirality associated with the hemiacetal containing carbon in compound 5 will be addressed later. Hence, once the equilibrium undergoes Fischer esterification in the presence of methanol, H2SO4, and heat, the major product will be compound 3.
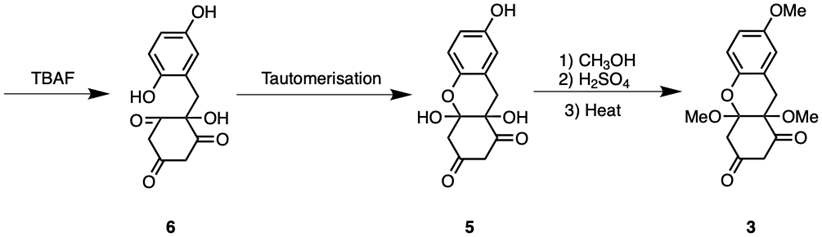
Figure 8. Synthesis of Uvarialeptone A & C Part 3.
Finally, compound 4 may be added with compound 3 along with LDA to form compound 24, which then undergoes keto/enol tautomerisation into uvarialeptone C. The tautomers are insignificant in this case, as they will freely interconvert, nonetheless, even if one specific tautomer were to be isolated. Teerapongpisan et al. presenting Uvarialeptone C as compound 2 as shown in Figure 9, suggests the specific enol form demonstrated is the most representative of the overall equilibrium [8].
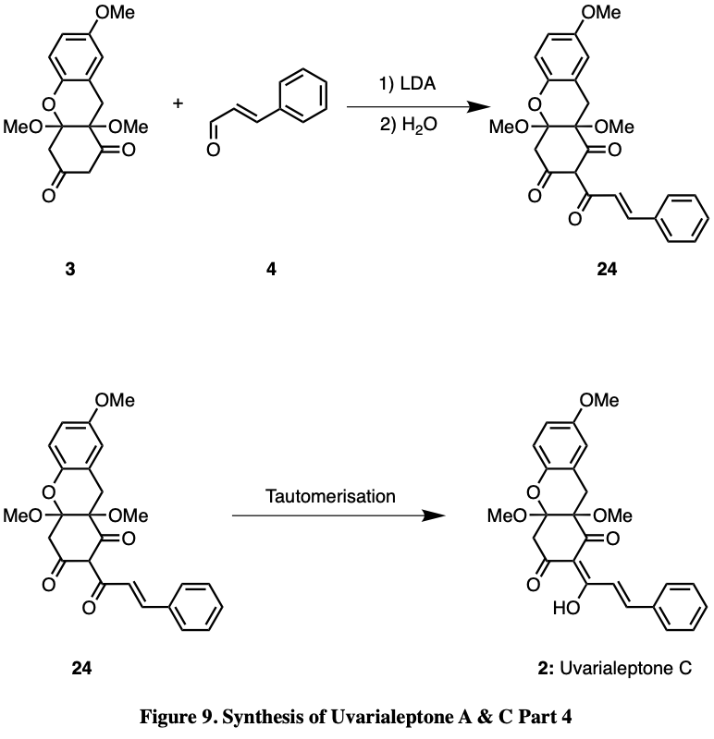
Figure 9. Synthesis of Uvarialeptone A & C Part 4.
As discussed in the retrosynthetic analysis, two uvarialeptone C molecules may undergo [2+2] cycloaddition catalysed by visible light, forming uvarialeptone A, as seen in Figure 10 [8].
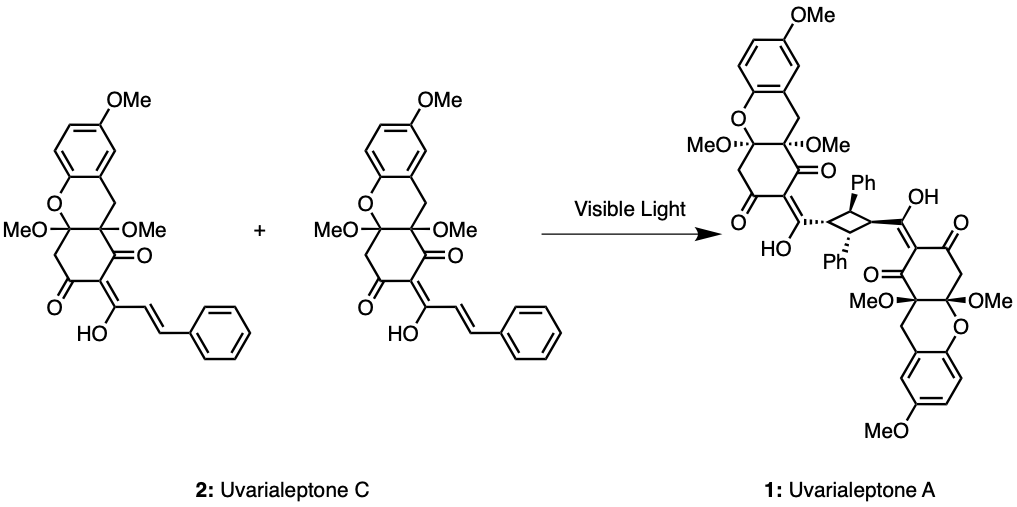
Figure 10. Synthesis of Uvarialeptone A & C Part 5.
4. Additional Remarks
4.1. Synthesis of Uvarialeptone B
Given the structural similarities between uvarialeptone A and B, much of the synthesis discussed above for uvarialeptone A also applies to uvarialeptone B, as their only difference exists between a hydrogen atom and a methoxy group. Additionally, like how uvarialeptone A is comprised of two uvarialeptone C molecules that underwent visible light catalysed [2+2] cycle addition, uvarialeptone is comprised of two 9a-O-methyloxymitrone molecules, as seen in Figure 11. Hence, the synthesis of uvarialeptone B also applies to 9a-O-methyloxymitrone.
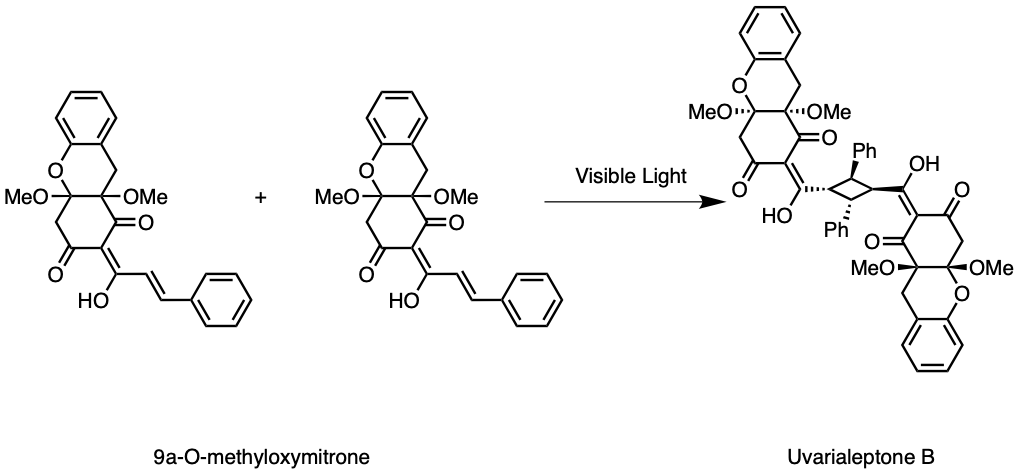
Figure 11. Uvarialeptone B Cycloaddition.
The only major difference between synthesising uvarialeptone A and B is that synthesising uvarialeptone B may require a Nagata reaction, as seen in Figure 12. Instead of starting with hydroquinone, the synthesis of uvarialeptone B starts with adding phenol to compounds 25 and 26. In the presence of 5 mol% ZrCl4, five equivalents of MgSO4 and left at moderate temperatures for more than 24 hours, compound 27 is formed. It is worth noting that the exact reaction conditions could be altered by using substitutes, though it is recommended to use ZrCl4 and MgSO4. It is also recommended to use 1,2-dichloroethane as the solvent and keep the system at 50°C, as such conditions were found to maximise the yield for more hindered aldehydes. Heating compound 27 disconnects the boron-containing six-member ring and destroys the phenyl group's aromaticity by shifting the position of a carbon-carbon π bond, as shown in compound 28. Adding tert-butylamine, borane and AlCl3 to compound 28 reduces the ketone attached to the six-member carbon ring back to alcohol and restores aromaticity, forming compound 29 [13].
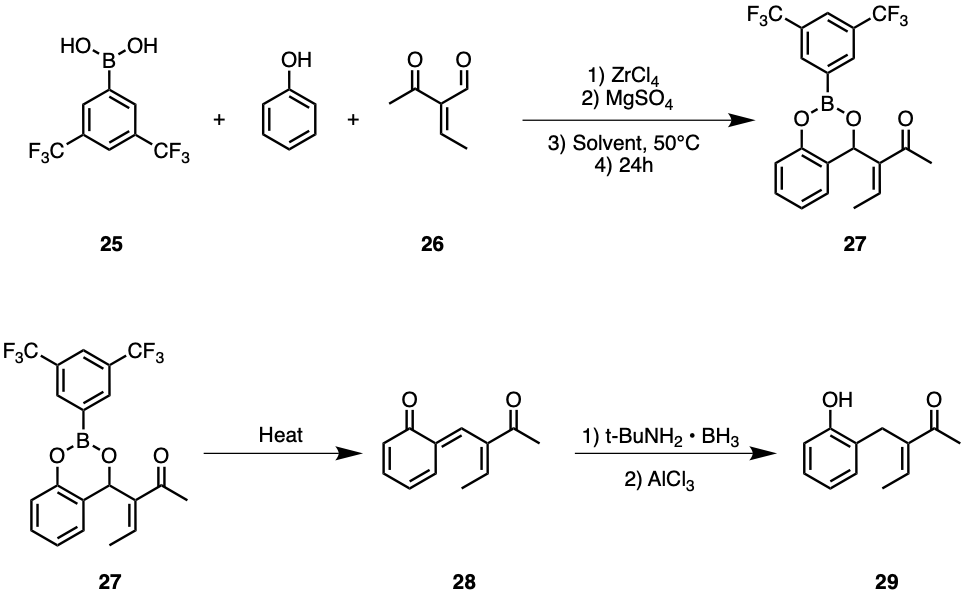
Figure 12. Synthesis of Uvarialeptone B.
4.2. Enantiopure Synthesis
The initial bioassay regarding uvarialeptones A and C was performed with enantiopure samples. Within the study, both chiral carbons (bridgehead carbons attached to methoxy groups) in uvarialeptone C are shown to be R enantiomers. The proposed synthesis may not generate an enantiopure sample of both uvarialeptones. Nevertheless, it may serve as a general outline. Additionally, investigation into a racemic mixture of both uvarialeptones may also prove fruitful.
Regardless, the chirality of either carbon bridgehead is controllable via enantioselective reactions. Regarding the chiral carbon connected to both a methoxy and a carbonyl, the implementation of Sharpless asymmetric dihydroxylation should guarantee products with enantiomeric excess up to 90% [14]. Using either (DHQD)2-PHAL or (DHQ)2-PHAL within the reaction will determine the enantiotopic configuration of the product [15]. Since the adjacent hydroxyl group generated via dihydroxylation will be oxidised, its chirality upon initial Sharpless asymmetric dihydroxylation is irrelevant.
As for the other chiral carbon in uvarialeptone C, an approach detailed by Borzecka et al. in the synthesis of enantiopure fluoromesonidazole could be similarly implemented [16]. Using alcohol hydrogenase from Rhodococcus ruber ADH and E. coli within the described reaction should reduce a ketone to a hydroxy in an R configuration. However, using ADH from Lactobacillus brevis, or LBADH, would reduce a ketone to a hydroxy in an S configuration [17]. Protecting groups should be added to all other ketones before undergoing this enantioselective process.
Alternatively, an Enders SAMP hydrazone alkylation reaction could also be used to control the stereochemistry of the second chiral centre. SAMP acts as a chiral auxiliary and ensures product stereoselectivity. It may be later removed via HCl [18]. Nevertheless, whether the enantioselectivity will behave expectedly when operating with ring/chain tautomerisation is unknown. Such an approach's results are less reliable than the previous proposal regarding the Sharpless asymmetric dihydroxylation.
5. Conclusion
The paper introduced prior research regarding Uvaria leptopoda derivatives and conducted retrosynthetic analysis on uvarialeptone A and C. A synthesis scheme of uvarialeptone A based on prior analysis results was proposed. The paper also predicts potential challenges regarding poor yields in the synthesis. The paper included additional relevant information regarding altering the proposed scheme for synthesising uvarialeptone B and strategies for synthesising enantiopure products. Nevertheless, the proposed scheme is purely theoretical. Further modifications to the synthesis scheme will be necessary to improve yields. Further practical investigations will also be necessary to evaluate the feasibility and efficiency of the proposed synthesis. Improvements to the proposed scheme may then be added after practical research.
References
[1]. Christopher, R. (2021). Plant species of the genus Uvaria: ethnobotanical uses, biological activities and phytochemistry. Natural Product Research, 36(11), 2946–2961. https://doi.org/10.1080/14786419.2021.1929972
[2]. Mfopa, A. N., Mbouna, C. D., Tchokouaha, L. R., Tchuente, M. A., Kouipou, R. M., Fokou, P. V., Kemgne, E. A., Kamkumo, R. G., & Boyom, F. F. (2017). in vitro and in vivo antiplasmodial activity of polyalthia suaveolens, uvaria angolensis and monodora tenuifolia (Annonaceae) extracts. International Journal of Biological and Chemical Sciences, 11(1), 118. https://doi.org/10.4314/ijbcs.v11i1.10
[3]. Ngoutane Mfopa, A., Corona, A., Eloh, K., Tramontano, E., Frau, A., Boyom, F. F., Caboni, P., & Tocco, G. (2017). Uvaria angolensis is a promising source of inhibitors of HIV-1 RT-associated RNA-dependent DNA polymerase and RNase H functions. Natural Product Research, 32(6), 640–647. https://doi.org/10.1080/14786419.2017.1332615
[4]. Auranwiwat, C., Wongsomboon, P., Thaima, T., Rattanajak, R., Kamchonwongpaisan, S., Willis, A. C., Laphookhieo, S., Pyne, S. G., & Limtharakul, T. (2019). Polyoxygenated cyclohexenes and their chlorinated derivatives from the leaves of uvaria cherrevensis. Journal of Natural Products, 82(1), 101–110. https://doi.org/10.1021/acs.jnatprod.8b00794
[5]. Auranwiwat, C., Rattanajak, R., Kamchonwongpaisan, S., Laphookhieo, S., Pyne, S. G., & Limtharakul, T. (2018). Four new C-benzyl flavonoids from the fruit of uvaria cherrevensis. Fitoterapia, 130, 198–202. https://doi.org/10.1016/j.fitote.2018.08.020
[6]. Meade, C. V., & Parnell, J. A. (2018). A revised taxonomy for Uvaria (Annonaceae) in continental Asia. Australian Systematic Botany, 31(4), 311–356. https://doi.org/10.1071/sb17051
[7]. Pailee, P., Ploypradith, P., Mahidol, C., Ruchirawat, S., & Prachyawarakorn, V. (2022). Dulcisenes C-E, polyoxygenated cyclohexenes, from Uvaria Dulcis Dunal and their cytotoxic activity. Phytochemistry, 202, 113298. https://doi.org/10.1016/j.phytochem.2022.113298
[8]. Teerapongpisan, P., Monkantha, T., Yimklan, S., Mah, S. H., Gunter, N. V., Promnart, P., Deachathai, S., Maneerat, T., Duangyod, T., Charoensup, R., Baka, A., Andersen, R. J., & Laphookhieo, S. (2024). Tetrahydroxanthene-1, 3(2H)-diones and Oxidized Hexadiene Derivatives from Uvaria leptopoda and Their Biological Activities. Journal of Natural Products, 87(6), 1611–1617. https://doi.org/10.1021/acs.jnatprod.4c00248
[9]. Ha, S., Lee, Y., Kwak, Y., Mishra, A., Yu, E., Ryou, B., & Park, C.-M. (2020). Alkyne–alkene [2 + 2] cycloaddition based on visible light photocatalysis. Nature Communications, 11(1). https://doi.org/10.1038/s41467-020-16283-9
[10]. Whiting, J. E., & Edward, J. T. (1971). Ring–chain tautomerism of Hydroxyketones. Canadian Journal of Chemistry, 49(23), 3799–3806. https://doi.org/10.1139/v71-635
[11]. Sasai, H., & Takizawa, S. (2012). 6.9 C–C bond formation: (aza) morita–baylis–hillman reaction. Comprehensive Chirality, 234–263. https://doi.org/10.1016/b978-0-08-095167-6.00609-1
[12]. Maloshitskaya, O. A., Sinkkonen, J., Alekseyev, V. V., Zelenin, K. N., & Pihlaja, K. (2005). A comparison of ring-chain tautomerism in heterocycles derived from 2-aminobenzenesulfonamide and anthranilamide. Tetrahedron, 61(30), 7294–7303. https://doi.org/10.1016/j.tet.2005.04.074
[13]. Zheng, H., & Hall, D. G. (2010). Zirconium-catalyzed nagata reaction for the synthesis of 2-aryl-1, 3, 2-aryldioxaborins via a mild three-component condensation of phenols, aldehydes, and boronic acid. Tetrahedron Letters, 51(32), 4256–4259. https://doi.org/10.1016/j.tetlet.2010.06.035
[14]. Mushtaq, A., Zahoor, A. F., Bilal, M., Hussain, S. M., Irfan, M., Akhtar, R., Irfan, A., Kotwica-Mojzych, K., & Mojzych, M. (2023). Sharpless asymmetric dihydroxylation: An impressive gadget for the synthesis of natural products: A Review. Molecules, 28(6), 2722. https://doi.org/10.3390/molecules28062722
[15]. Ager, D. (2012). 3.9 alkaloid derived auxiliaries: Cinchona alkaloids and derivatives. Comprehensive Chirality, 223–247. https://doi.org/10.1016/b978-0-08-095167-6.00309-8
[16]. Bastrakov, M., & Starosotnikov, A. (2022). Recent progress in the synthesis of drugs and bioactive molecules incorporating nitro(het)arene core. Pharmaceuticals, 15(6), 705. https://doi.org/10.3390/ph15060705
[17]. Borzęcka, W., Lavandera, I., & Gotor, V. (2013). Biocatalyzed synthesis of both Enantiopure fluoromisonidazole antipodes. Tetrahedron Letters, 54(37), 5022–5025. https://doi.org/10.1016/j.tetlet.2013.07.013
[18]. Job, A., Janeck, C. F., Bettray, W., Peters, R., & Enders, D. (2002). The SAMP-/ramp-hydrazone methodology in asymmetric synthesis. Tetrahedron, 58(12), 2253–2329. https://doi.org/10.1016/s0040-4020(02)00080-7
Cite this article
Ma,H. (2025). Retrosynthesis and Synthesis Proposal of Uvarialeptones. Theoretical and Natural Science,78,138-147.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Christopher, R. (2021). Plant species of the genus Uvaria: ethnobotanical uses, biological activities and phytochemistry. Natural Product Research, 36(11), 2946–2961. https://doi.org/10.1080/14786419.2021.1929972
[2]. Mfopa, A. N., Mbouna, C. D., Tchokouaha, L. R., Tchuente, M. A., Kouipou, R. M., Fokou, P. V., Kemgne, E. A., Kamkumo, R. G., & Boyom, F. F. (2017). in vitro and in vivo antiplasmodial activity of polyalthia suaveolens, uvaria angolensis and monodora tenuifolia (Annonaceae) extracts. International Journal of Biological and Chemical Sciences, 11(1), 118. https://doi.org/10.4314/ijbcs.v11i1.10
[3]. Ngoutane Mfopa, A., Corona, A., Eloh, K., Tramontano, E., Frau, A., Boyom, F. F., Caboni, P., & Tocco, G. (2017). Uvaria angolensis is a promising source of inhibitors of HIV-1 RT-associated RNA-dependent DNA polymerase and RNase H functions. Natural Product Research, 32(6), 640–647. https://doi.org/10.1080/14786419.2017.1332615
[4]. Auranwiwat, C., Wongsomboon, P., Thaima, T., Rattanajak, R., Kamchonwongpaisan, S., Willis, A. C., Laphookhieo, S., Pyne, S. G., & Limtharakul, T. (2019). Polyoxygenated cyclohexenes and their chlorinated derivatives from the leaves of uvaria cherrevensis. Journal of Natural Products, 82(1), 101–110. https://doi.org/10.1021/acs.jnatprod.8b00794
[5]. Auranwiwat, C., Rattanajak, R., Kamchonwongpaisan, S., Laphookhieo, S., Pyne, S. G., & Limtharakul, T. (2018). Four new C-benzyl flavonoids from the fruit of uvaria cherrevensis. Fitoterapia, 130, 198–202. https://doi.org/10.1016/j.fitote.2018.08.020
[6]. Meade, C. V., & Parnell, J. A. (2018). A revised taxonomy for Uvaria (Annonaceae) in continental Asia. Australian Systematic Botany, 31(4), 311–356. https://doi.org/10.1071/sb17051
[7]. Pailee, P., Ploypradith, P., Mahidol, C., Ruchirawat, S., & Prachyawarakorn, V. (2022). Dulcisenes C-E, polyoxygenated cyclohexenes, from Uvaria Dulcis Dunal and their cytotoxic activity. Phytochemistry, 202, 113298. https://doi.org/10.1016/j.phytochem.2022.113298
[8]. Teerapongpisan, P., Monkantha, T., Yimklan, S., Mah, S. H., Gunter, N. V., Promnart, P., Deachathai, S., Maneerat, T., Duangyod, T., Charoensup, R., Baka, A., Andersen, R. J., & Laphookhieo, S. (2024). Tetrahydroxanthene-1, 3(2H)-diones and Oxidized Hexadiene Derivatives from Uvaria leptopoda and Their Biological Activities. Journal of Natural Products, 87(6), 1611–1617. https://doi.org/10.1021/acs.jnatprod.4c00248
[9]. Ha, S., Lee, Y., Kwak, Y., Mishra, A., Yu, E., Ryou, B., & Park, C.-M. (2020). Alkyne–alkene [2 + 2] cycloaddition based on visible light photocatalysis. Nature Communications, 11(1). https://doi.org/10.1038/s41467-020-16283-9
[10]. Whiting, J. E., & Edward, J. T. (1971). Ring–chain tautomerism of Hydroxyketones. Canadian Journal of Chemistry, 49(23), 3799–3806. https://doi.org/10.1139/v71-635
[11]. Sasai, H., & Takizawa, S. (2012). 6.9 C–C bond formation: (aza) morita–baylis–hillman reaction. Comprehensive Chirality, 234–263. https://doi.org/10.1016/b978-0-08-095167-6.00609-1
[12]. Maloshitskaya, O. A., Sinkkonen, J., Alekseyev, V. V., Zelenin, K. N., & Pihlaja, K. (2005). A comparison of ring-chain tautomerism in heterocycles derived from 2-aminobenzenesulfonamide and anthranilamide. Tetrahedron, 61(30), 7294–7303. https://doi.org/10.1016/j.tet.2005.04.074
[13]. Zheng, H., & Hall, D. G. (2010). Zirconium-catalyzed nagata reaction for the synthesis of 2-aryl-1, 3, 2-aryldioxaborins via a mild three-component condensation of phenols, aldehydes, and boronic acid. Tetrahedron Letters, 51(32), 4256–4259. https://doi.org/10.1016/j.tetlet.2010.06.035
[14]. Mushtaq, A., Zahoor, A. F., Bilal, M., Hussain, S. M., Irfan, M., Akhtar, R., Irfan, A., Kotwica-Mojzych, K., & Mojzych, M. (2023). Sharpless asymmetric dihydroxylation: An impressive gadget for the synthesis of natural products: A Review. Molecules, 28(6), 2722. https://doi.org/10.3390/molecules28062722
[15]. Ager, D. (2012). 3.9 alkaloid derived auxiliaries: Cinchona alkaloids and derivatives. Comprehensive Chirality, 223–247. https://doi.org/10.1016/b978-0-08-095167-6.00309-8
[16]. Bastrakov, M., & Starosotnikov, A. (2022). Recent progress in the synthesis of drugs and bioactive molecules incorporating nitro(het)arene core. Pharmaceuticals, 15(6), 705. https://doi.org/10.3390/ph15060705
[17]. Borzęcka, W., Lavandera, I., & Gotor, V. (2013). Biocatalyzed synthesis of both Enantiopure fluoromisonidazole antipodes. Tetrahedron Letters, 54(37), 5022–5025. https://doi.org/10.1016/j.tetlet.2013.07.013
[18]. Job, A., Janeck, C. F., Bettray, W., Peters, R., & Enders, D. (2002). The SAMP-/ramp-hydrazone methodology in asymmetric synthesis. Tetrahedron, 58(12), 2253–2329. https://doi.org/10.1016/s0040-4020(02)00080-7