







1. Introduction
Transcriptome is the collection of all transcripts produced by a particular species or cell type. Transcriptome research can study gene function and gene structure from the overall level, reveal the molecular mechanism of specific biological processes and disease occurrence, and has been widely used in basic research, clinical diagnosis drug discovery and development, etc. [1] RNA-seq (RNA sequencing) is a transcriptome sequencing technology, which is the use of high-throughput sequencing technology for sequencing analysis, reflecting the expression level of mRNA, small RNA, noncoding RNA, or some of them. RNA-seq (RNA sequencing) is a technology that uses high-throughput sequencing to reflect the expression level of mRNA, small RNA, noncoding RNA, or some of them.
For a complete transcriptome data analysis process, total mRNA is extracted from the study samples and subjected to library construction and sequencing, quality control is performed on the downstream data obtained, and a suitable comparison tool is selected to compare the processed reads to the reference genome. Finally, the read counts for each gene were extracted from the comparison results using the tool to generate a count matrix of gene expression. The gene expression counts are then statistically analyzed to identify differential genes. All of this places higher demands on the researcher.
In recent years, transcriptome sequencing has gained unprecedented momentum following the popularity of high-throughput sequencing. Depending on the direction of the research content, the accuracy, speed, and cost requirements vary, and researchers need to weigh up elements including the specific sequencing methodology process to be adopted, the type of samples, and the desired analytical results, as well as the current state of genomic research and the resources available for computational data processing. Because of the complexity and diversity of the issues involved, it is critical to find an optimal workflow to optimize the different aspects involved in RNA-seq analysis based on cost and performance requirements. Meanwhile, with the development of third-generation sequencing technology, nanopore sequencing technology, which is more advantageous in terms of read length and efficiency, is also playing an increasing role in transcriptome research.
2. Overview and Development of Sequencing Technology
In 1977, Walter Gilbert and Frederick Sanger invented the first sequencing machine and applied it to sequence the first genome, that of bacteriophage X174, which is 5375 bases long in total. This marked the beginning of humanity's ability to explore the genetic essence of life, ushering the life sciences into the era of genomics. Over the past four decades, sequencing technology has made tremendous advancements, evolving from the first generation to the third generation. The completion of the Human Genome Project has enabled researchers to move beyond the identification and annotation of genetic information at the chromosomal level to studying the genetic information in mRNA and transcripts. New transcripts and expression regulatory mechanisms have been continuously discovered, and further research using newly annotated transcripts has been ongoing. Researchers have been continuously accumulating data while mapping their regulatory networks, providing new insights into diseases and growth and development. Additionally, in recent years, the continuous improvement of emerging technologies such as artificial intelligence has promoted further development of sequencing technology.
2.1. First-Generation Sequencing (FGS)
In 1977, Maxam and Gilbert began using chemical degradation to sequence DNA, and around the same time, Sanger introduced dideoxyribonucleotide triphosphates (ddNTPs), leading to the development of the dideoxy chain termination method, which significantly enhanced the efficiency and accuracy of DNA sequencing. DNA sequencing technology based on both the dideoxy chain termination method and chemical degradation is referred to as First-Generation Sequencing (FGS). The Sanger sequencing method involves the incorporation of a small number of ddNTPs, randomly terminating the synthesis of the nucleic acid chain at a specific ddNTP. High-resolution denaturing gel electrophoresis is then used to differentiate nucleic acids at different positions. First-generation sequencing is considered the gold standard in nucleic acid sequencing due to its accuracy. However, FGS is characterized by high costs, long duration, and low throughput [2]. Degradation of samples or primer failure can reduce sequencing quality. For samples containing special nucleic acid structures, such as repetitive sequences, palindromic sequences, hairpin structures, GC-rich regions, and AT-rich regions, the quality of Sanger sequencing is also poor.
2.2. Second-Generation Sequencing (NGS)
High-throughput sequencing (HTS), also known as Next Generation Sequencing (NGS), is a revolutionary advancement over traditional Sanger sequencing. It allows for the sequencing of tens of thousands to millions of nucleic acid molecules simultaneously. Mature second-generation sequencing platforms include Roche-454 Life Sciences (emulsion PCR), Illumina Method (bridge PCR), Solid Method (emulsion PCR using magnetic beads), and Ion Torrent Method (emulsion PCR combined with pH ion changes). The advent of second-generation sequencing has enabled researchers to generate vast amounts of data in a short period. Additionally, the fragments sequenced by second-generation sequencing are shorter. In terms of accuracy, due to the introduction of PCR technology, a low mismatch rate during synthesis may still lead to sequence alignment errors [3]. Furthermore, enzymatic limitations of DNA polymerases may result in incorrect sequence reads in repetitive sequences (including homopolymers) and CG-rich regions. During data processing based on second-generation sequencing, smaller fragments need to be assembled and paired. In the absence of a reference genome, it can be difficult to assemble the correct genome due to repetitive sequences or fragments that appear at multiple loci in the genome, making it challenging to map specific short sequences to specific loci.
2.3. Third-Generation Sequencing (TGS)
Neither first- nor second-generation sequencing technologies target individual DNA molecules. The emergence of third-generation sequencing technology has made it possible to sequence individual DNA or RNA sequences without prior reverse transcription or amplification. Third-generation sequencing, characterized by long read lengths and independence from transcription and amplification, provides new tools for research in single-cell genomics and alternative splicingomics [4]. Additionally, third-generation sequencing technologies, such as nanopore sequencing, can be combined with deep learning techniques for functions like protein methylation recognition. Currently, the error rate of third-generation sequencing technology is still relatively high. In 2019, PacBio introduced high-fidelity (HiFi) reads with lengths of 10-20 kb and an error rate below 0.5% [5]. However, single-base mismatches in third-generation sequencing can be addressed by increasing sequencing depth. Compared to the first two generations of sequencing technology, third-generation sequencing is more costly but better at recognizing repetitive sequences.
3. Bulk RNA-Seq
Bulk RNA sequencing, in contrast to single cell sequencing technology, targets populations of cells. Sequencing the Total RNA from certain tissues, organs, or cell populations, reflects the overall gene expression level of the studied cell population [6]. Bulk RNA-Seq is widely used to compare gene expression differences between different tissues or individuals, as well as to explore gene expression changes under specific conditions. In disease research, Bulk RNA-Seq can assist researchers in identifying genes and pathways associated with diseases, providing clues for disease diagnosis and treatment [7].
3.1. Upstream Data Processing
The overall environment has been configured using Miniconda, with all necessary tools loaded within this environment. The core objective of upstream data analysis and organization is to obtain matrices for individual reads. Firstly, raw data (fastq format) is obtained from sequencing instruments. Subsequently, the Trimmomatic software package removes primer sequences from the raw data based on the upstream and downstream primer sequences specific to different sequencing platforms, resulting in clean data. Next, gene sequences and gene annotation files (fasta and gtf formats) for the target organism are obtained from authoritative websites such as UCSC and Gencode. Using the Hisat2 software package, the cleaned data is aligned with the gene sequences to generate alignment results (sam format). Then, the sam files are converted into the more efficient bam format using the Samtools tool. FeatureCounts are employed to count the alignment results, generating detailed count results and count summary files. Finally, the detailed alignment results are converted into tsv format, and the count results are renamed further simplified, and modified [8].
3.2. Downstream Data Processing and Analysis Workflow
In the downstream data processing and analysis phase, we use the "DESeq2" package in R to read the detailed alignment results (tsv format). Firstly, a grouping table is constructed to clarify the classification relationships between samples. Next, data with excessively low counts is filtered out to improve the accuracy of the analysis. Then, PCA (Principal Component Analysis) is performed on the obtained results to reveal potential relationships between samples. Finally, volcano plots are drawn to visually display changes in gene expression levels and their statistical significance. Subsequent analysis will involve Go analysis and KEGG analysis to interpret pathway alterations [9].
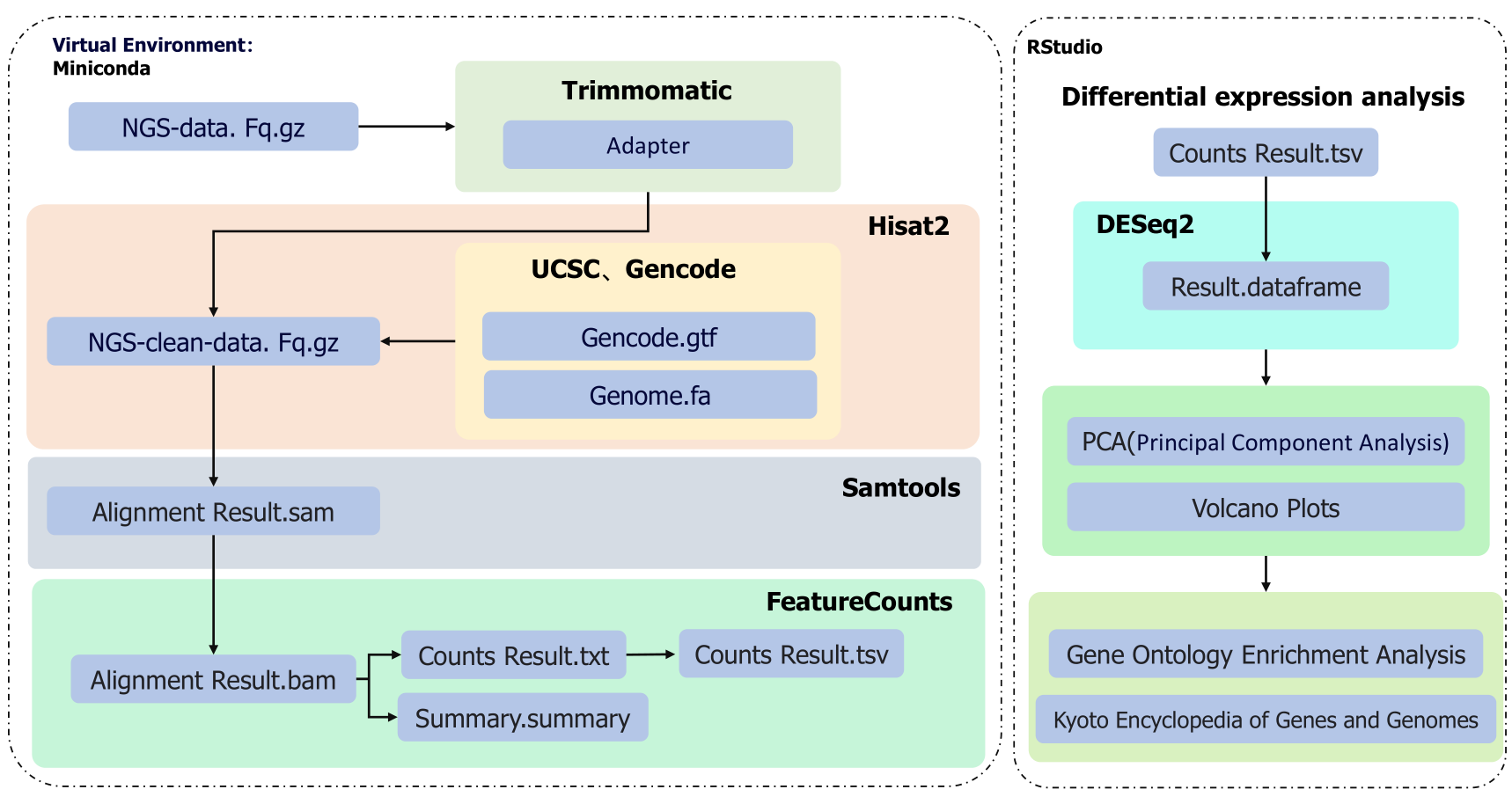
Figure 1: Flow Chart of Bulk RNA-Seq
4. Oxford Nanopore Technologies
Oxford Nanopore Technologies (ONT) sequencing technology is currently the mainstream third-generation sequencing platform. It completes sample pretreatment by adding adapter sequences and adaptive motor proteins to the target nucleic acid sequences [10]. In a container filled with electrolytes, a molecular membrane embedded with nanopore proteins is placed, and only nanopores on the membrane allow ions to pass through. Motor proteins pull the nucleic acid sequence close and guide the motor protein-attached chain to unzip and pass through the nanopore protein at a relatively stable rate. When different nucleic acid sequences pass through the nanopore protein, they will cause a potential difference between the two solutions stably controlled by an external power supply. After transmitting the information of potential difference changes to the processing center, combined with deep learning methods, the nucleic acid sequence can be obtained in real time.
Currently, the ONT platform is mainly used in direct cDNA sequencing, PCR-cDNA sequencing, and RNA sequencing. Besides simple sequencing, it is also applied to differential gene expression (DGE) studies. DGE studies are primarily completed using RNA-seq, which reflects different splice variants and gene expression profiles. Currently, DGE work based on RNA-seq is mainly developed on the Illumina platform. On the Illumina platform, high-throughput sequencing of RNA is completed through steps such as conventional cell lysis to obtain cell lysate, RNA extraction, RNA enrichment, removal of ribosomal RNA, adapter ligation sequencing, etc [11]. Increasing sequencing depth can improve sequencing accuracy to a certain extent. With the continuous development of the ONT platform, researchers have continuously enhanced the long-read sequencing strategy for the ONT platform. Compared with the short-read high-throughput results of the Illumina platform, the long-read sequencing results of the ONT platform provide splice variant identification and direct acquisition of complete mRNA sequences. Direct sequencing of DNA on the ONT platform can reveal certain base modifications. In addition, the ONT platform is also applied in current fields such as single-cell sequencing. The ONT platform is currently the only platform that supports both DNA and RNA sequencing. The main issue facing the ONT platform is that the error rate of ONT sequencing results after PCR combined with PCR is orders of magnitude higher than that of second-generation sequencing on the Illumina platform, which is related to errors and methods in the PCR process. However, the primary purpose of research institutions using the ONT platform is to obtain full-length sequences, and individual base errors are acceptable.
5. Discussion
Currently, there is no perfect platform that suits all types of experiments, as second-generation and third-generation sequencing technologies each have their strengths and weaknesses. In recent years, long-read sequencing strategies have emerged as hot topics. With the support of short-read high-depth strategies for basic data, researchers have begun to realize that the information obtained from long-read sequencing using platforms like ONT can further explain transcriptome variations, particularly in the context of alternative splicing. The ONT platform has opened the door to understanding RNA epigenetic modifications, further advancing our knowledge of genetics and variations. However, platforms like ONT still face issues such as high error rates, high costs, and low throughput. In recent years, some teams have proposed high-fidelity long-read sequencing strategies and improvements in correct recognition rates through deep learning [12]. Over decades of development, sequencing technology has successfully been applied in fields such as disease diagnosis, treatment, growth and development identification, and drug research and development. With continuous interdisciplinary integration, traditional sequencing technology may cross-collaborate with other disciplines to develop a more efficient, economical, and accurate unified sequencing platform.
6. Conclusion
After more than 100 years of development, sequencing technology has matured significantly. The accuracy of first-generation sequencing technology has made it the gold standard in sequencing, but its low throughput has rendered it insufficient for certain current sequencing demands. The emergence of second-generation sequencing technology addressed the low throughput issue of first-generation technology, enabling high-throughput and high-depth sequencing, although it also faced the problem of short reads. The advent of third-generation sequencing technology compensated for the short reads issue of second-generation technology, becoming the first sequencing technology capable of long reads and high throughput. However, it also has a higher error rate compared to first and second-generation sequencing technologies, although this error can sometimes be corrected by increasing depth. With the development of sequencing technology, its applications have expanded into various fields, evolving from simple gene information analysis to transcript information analysis and differential expression analysis, leading to advancements in areas such as Bulk RNA-seq, single-cell sequencing, spliceosome identification, and epigenetic research. Among these, Bulk RNA-seq identifies differential gene expression between different cell populations through gene analysis, revealing disease etiology, analyzing treatment plans, guiding drug development, and studying biological regulatory processes, among others. The ONT technology in third-generation sequencing provides opportunities to identify methylation modifications, opening new avenues for epigenetic research, while longer reads allow for the acquisition of complete transcript information and the discovery of new alternative splicing patterns. The applications across various fields rely on the sharing and accumulation of global annotation information, and the continuously accumulating libraries make the development of new information relatively easier. However, there is currently a lack of research methods specifically targeting highly repetitive sequences, especially in centromeric regions, and there is a shortage of unified analysis platforms that can meet various sequencing needs simultaneously. The introduction of new technologies from computer science, physics, and chemistry has also brought new possibilities for the development of sequencing technology.
References
[1]. Segel, M. et al. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science 373, 882-+ (2021).
[2]. Dorado, G., Gálvez, S., Rosales, T.E., Vásquez, V.F. & Hernández, P. Analyzing Modern Biomolecules: The Revolution of Nucleic-Acid Sequencing - Review. Biomolecules 11 (2021).
[3]. Jia, H.X., Tan, S.J. & Zhang, Y.E. Chasing Sequencing Perfection: Marching Toward Higher Accuracy and Lower Costs. Genomics Proteomics & Bioinformatics 22 (2024).
[4]. Glinos, D.A. et al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 608, 353-+ (2022).
[5]. Li, H. & Durbin, R. Genome assembly in the telomere-to-telomere era. ArXiv (2023).
[6]. Li, X.M. & Wang, C.Y. From bulk, single-cell to spatial RNA sequencing. International Journal of Oral Science 13 (2021).
[7]. Thind, A.S. et al. Demystifying emerging bulk RNA-Seq applications: the application and utility of bioinformatic methodology. Briefings in Bioinformatics 22 (2021).
[8]. Yu, X., Abbas-Aghababazadeh, F., Chen, Y.A. & Fridley, B.L. Statistical and Bioinformatics Analysis of Data from Bulk and Single-Cell RNA Sequencing Experiments. Methods in molecular biology (Clifton, N.J.) 2194, 143-175 (2021).
[9]. Thomas, P.D., Mi, H.Y. & Lewis, S. Ontology annotation: mapping genomic regions to biological function. Current Opinion in Chemical Biology 11, 4-11 (2007).
[10]. Dorey, A. & Howorka, S. Nanopore DNA sequencing technologies and their applications towards single-molecule proteomics. Nature Chemistry 16, 314-334 (2024).
[11]. Stark, R., Grzelak, M. & Hadfield, J. RNA sequencing: the teenage years. Nature Reviews Genetics 20, 631-656 (2019).
[12]. Farrow, E. et al. Case of CLPB deficiency solved by HiFi long read genome sequencing and RNAseq. American Journal of Medical Genetics Part A 191, 2908-2912 (2023).
Cite this article
Zhang,H. (2025). Development and Application of Transcript Sequencing. Theoretical and Natural Science,89,1-6.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 3rd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Segel, M. et al. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science 373, 882-+ (2021).
[2]. Dorado, G., Gálvez, S., Rosales, T.E., Vásquez, V.F. & Hernández, P. Analyzing Modern Biomolecules: The Revolution of Nucleic-Acid Sequencing - Review. Biomolecules 11 (2021).
[3]. Jia, H.X., Tan, S.J. & Zhang, Y.E. Chasing Sequencing Perfection: Marching Toward Higher Accuracy and Lower Costs. Genomics Proteomics & Bioinformatics 22 (2024).
[4]. Glinos, D.A. et al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 608, 353-+ (2022).
[5]. Li, H. & Durbin, R. Genome assembly in the telomere-to-telomere era. ArXiv (2023).
[6]. Li, X.M. & Wang, C.Y. From bulk, single-cell to spatial RNA sequencing. International Journal of Oral Science 13 (2021).
[7]. Thind, A.S. et al. Demystifying emerging bulk RNA-Seq applications: the application and utility of bioinformatic methodology. Briefings in Bioinformatics 22 (2021).
[8]. Yu, X., Abbas-Aghababazadeh, F., Chen, Y.A. & Fridley, B.L. Statistical and Bioinformatics Analysis of Data from Bulk and Single-Cell RNA Sequencing Experiments. Methods in molecular biology (Clifton, N.J.) 2194, 143-175 (2021).
[9]. Thomas, P.D., Mi, H.Y. & Lewis, S. Ontology annotation: mapping genomic regions to biological function. Current Opinion in Chemical Biology 11, 4-11 (2007).
[10]. Dorey, A. & Howorka, S. Nanopore DNA sequencing technologies and their applications towards single-molecule proteomics. Nature Chemistry 16, 314-334 (2024).
[11]. Stark, R., Grzelak, M. & Hadfield, J. RNA sequencing: the teenage years. Nature Reviews Genetics 20, 631-656 (2019).
[12]. Farrow, E. et al. Case of CLPB deficiency solved by HiFi long read genome sequencing and RNAseq. American Journal of Medical Genetics Part A 191, 2908-2912 (2023).