







1. Research Background
Catechol (C6H6O2) is a common intermediate in the degradation of aromatic compounds, widely used in industries such as flavoring, fragrances, dyes, pharmaceuticals, and pesticides [1]. Annually, around 25,000 tons of catechol are improperly discharged into the environment [2]. As a toxic compound, catechol can irritate the skin and respiratory system, and long-term exposure may affect the central nervous system. The IARC classified catechol as a Group 2B carcinogen in 1999 [3].
Catechol’s solubility in water leads to significant water and soil pollution, accumulating in the food chain and impacting human and animal health, as well as disrupting ecosystem functions [4]. Due to its stable structure, catechol degrades slowly in nature, making it essential to explore efficient degradation methods. This study aims to identify effective pathways to reduce catechol's environmental and health impacts.
Current degradation methods include physical, chemical, and biological approaches. Physical methods, such as radiation ionization and pyrolysis, are costly and not widely applicable. Chemical methods like Fenton’s reaction and electrochemical oxidation also have limitations in complexity and cost [5-8]. Biological degradation, on the other hand, offers a low-cost, efficient alternative that completely breaks down catechol into CO2 and H2O with no secondary pollution.
Biological methods rely on microorganisms or expressed enzymes to degrade catechol. Commonly used microbes include Rhizobium, Saccharomyces, and Pseudomonas [9]. Key enzymes involved are catechol 1,2-dioxygenase (C12O) and catechol 2,3-dioxygenase (C23O), which catalyze the cleavage of the aromatic ring [10,11]. However, the growth of these microbes and enzyme activity can be impacted by environmental factors, limiting their practical application.
Immobilizing proteins or microbes onto surfaces enhances stability and efficiency. For example, immobilizing enzymes in calcium alginate hydrogels improves stability and increases their optimal temperature [12]. Similarly, surface display systems using microbial cells can bypass protein purification, reducing costs and improving tolerance to harsh conditions [13]. The Bacillus subtilis spore surface display system takes advantage of the spore’s resilience, offering a stable platform for protein immobilization. This system also reduces the need for purification steps and can operate under extreme conditions like high temperatures and organic solvents. The GRAS status of Bacillus subtilis spores further expands their potential applications, particularly in probiotic research [14].
The Bacillus subtilis spore system offers several advantages over other display systems. Since spores do not rely on membrane transport systems, even difficult-to-secrete proteins can be displayed. Additionally, spores show remarkable resilience, maintaining biological activity even after long storage. This system avoids the limitations of other display methods, such as post-translational modifications, and ensures better safety, stability, and cost-effectiveness [14].
In this study, I applied the Bacillus subtilis spore surface display system to immobilize C12O and C23O, key enzymes for catechol degradation. Using cotG as the anchoring protein, both enzymes were successfully displayed on the spore surface. Western blotting and immunofluorescence confirmed the successful expression of these enzymes. Enzyme activity assays revealed that both C12O and C23O on spores retained catechol-degrading activity, with broader pH and temperature tolerance compared to other reported enzymes. The spores also exhibited high resistance to methanol, confirming the stability and potential for use in harsh environments.
2. Research Focus
This study investigates the application of the Bacillus subtilis spore surface display system in catechol degradation. The goal is to explore an efficient biological degradation method for catechol, an environmental pollutant with significant health risks. The main objectives of this research were:
1. Engineering Recombinant Bacillus subtilis: The genes encoding C12O and C23O were fused with the cotG anchoring protein gene of Bacillus subtilis, enabling stable enzyme display on the spore surface. This system eliminates the need for traditional protein purification, reducing production costs.
2. Gene Cloning and Transformation: The genes for cotG, C12O, and C23O were amplified by PCR, cloned into expression vectors, and transferred to Bacillus subtilis for spore surface display.
3. Protein Display Confirmation: Western blot and immunofluorescence were used to confirm the successful display of C12O and C23O on the spore surface, demonstrating their stability and activity under extreme conditions.
4. Enzyme Activity Analysis: The activity of the displayed enzymes was tested across a range of pH and temperature conditions. The enzymes showed high stability and activity even under harsh conditions, confirming the potential of the spore surface display system for real-world applications in environmental pollution control.
Overall, this study demonstrates the feasibility of using the Bacillus subtilis spore surface display system to efficiently degrade catechol, offering a novel approach for environmental remediation and advancing biotechnological applications.
3. Research Methods
3.1. Experimental Materials
3.1.1. Strains and Plasmids
Strains: Escherichia coli DH5α; Bacillus subtilis DB104.
Plasmid: pHY300PLK.
3.1.2. Primers
Primer name | Primer sequence(5’—3’) |
cotG-phy-F | GGAAAAACGCTTTGCCCAGTGTCCCTAGCTCCGAGAAAAA |
cotG-phy-R | CAGATCTCTAGAAGCTTTTTGTATTTCTTTTTGACTACCCA |
pHY300plk-JD-F | GTTTCGCCACCACTGATTTGAGC |
pHY300plk-JD-R | TGTCATTAGTTGGCTGGTTACCT |
C12O-EcoRI-F | CCCAAGCTTGGTGGCGGTAGCGGCGGTGGCAG |
C12O-EcoRI-R | GGAATTCTCAGTGGTGGTGGTGGTGGTGGGTCAGCACGGTCATGAATCGT |
C23O-EcoRI-F | CCCAAGCTTGGTGGCGGTAGCGGCGGTGGCAG |
C23O-EcoRI-R | GGAATTCTCAGTGGTGGTGGTGGTGGTGGGTCAGCACGGTCAGGAAC |
3.1.3. Main Experimental Instruments
PCR Amplifier (Eppendorf, X50s); Constant Temperature Shaker (Zhichu, ZQZY-CR8); Protein Electrophoresis Equipment (BioRad); Gel Imaging System (GE Healthcare, AI600); Confocal Laser Scanning Microscope (Olympus, FV4000); Fluorescence Spectrophotometer (BioTek, FLY800).
3.1.4. Main Reagents
A. Plasmid Construction
PCR Enzyme (Takara); FastDigest Hind III, EcoRI (Thermo Fisher); Hieff Clone® PlusOne Step Cloning Kit (Yeasen); DH5α Competent Cells (Beyotime); Ampicillin (Beyotime); T4 DNA Ligase (NEB).
B. Strain Culture and Spore Induction
Trypsin (Sigma); Yeast Extract (Sigma); NaCl (Adamas); Bacto Nutrient Broth (Macklin); KCl, MgSO4, NaOH, and other chemicals were purchased from Adamas.
C. Protein Immunoblotting
Lysozyme, DTT (Beyotime); PAGE Gel Fast Preparation Kit (Epizyme Biotech); PVDF Membrane (Millipore); BSA (Sigma); TWEEN 20, Tris, and other chemicals were purchased from Adamas; Anti-6xHis Tag Antibody (Abcam); Rabbit Anti-Mouse IgG H&L (HRP) (Abcam); ECL Detection Kit (Beyotime).
D. Immunofluorescence Experiment
4% Paraformaldehyde Fixative (Beyotime); Goat Anti-Mouse IgG H&L (FITC) (Abcam); ProLong™ Glass Antifade Mountant (Invitrogen).
E. Enzyme Activity Analysis
Catechol, Methanol, and Diethyl Ether purchased from Sigma; Trypsin and Proteinase purchased from Beyotime.
3.1.5. Main Culture Media
LB Medium: 1% (W/V) Tryptone; 0.5% (W/V) Yeast Extract; 1% (W/V) NaCl. For solid medium, add 1.5% (W/V) Agar.
DSM Medium: 8 g Bacto Nutrient Broth; 10 mL 10% (W/V) KCl; 10 mL 1.2% (W/V) MgSO4·7H2O; 1.5 mL (pH to 7.6) 1 M NaOH; 1 mL 1M Ca(NO3)2; 1 mL 0.01 M MnCl2; 1 mM FeSO4.
3.2. Experimental Methods
3.2.1. Gene Amplification
Table 1: PCR amplification system
2×PCR mix | 25 μL |
Primer 1 | 2 μL |
Primer 2 | 2 μL |
DNA | 2 μL |
ddH2O | Make up to 50 μL |
Total | 50 μL |
PCR amplification procedure: First, pre-denature at 94°C for 5 minutes, followed by 30 seconds of denaturation at 94°C, 30 seconds of annealing at 55°C, and 30 seconds of extension at 72°C, repeated for 30 cycles. Finally, store at 10°C until the PCR product is collected.
3.2.2. Plasmid Digestion
Table 2: Digestion system
10×fast digest buffer | 5 μL |
Endonuclease 1 | 1 μL |
Plasmids | 20 μL |
ddH2O | Make up to 50 μL |
Total | 50 μL |
Digestion procedure: Incubate at 37°C for 1-3 hours.
3.2.3. Seamless Cloning Reaction
Table 3: Seamless cloning system
5×infusion cloing mix | 2 μL |
Carrier recovery product | 2 μL |
Fragment recovery products | 6 μL |
Total | 10 μL |
Seamless cloning procedure: Mix the reaction and incubate on ice for 30 minutes.
3.2.4. Transformation of Escherichia coli
First, thaw 100 μL of DH5α competent cells on ice, then add 10 μL of ligation product, gently mix by flicking the tube, and incubate on ice for 30 minutes. Next, heat shock the mixture in a 42°C water bath for 30 seconds, and immediately transfer to ice for 2 minutes (do not shake the tube during this step). Add 900 μL LB liquid medium to the competent cells and incubate at 37°C with shaking at 200 rpm for 1 hour. Then, centrifuge at 5000 rpm for 5 minutes, discard the supernatant, and resuspend the remaining 100 μL culture in the pellet. Spread the resuspended cells evenly onto LB solid plates containing ampicillin. Seal the plates with parafilm and invert them in a 37°C incubator, incubating for 12-16 hours until single colonies appear.
3.2.5. Colony PCR Identification
Table 4: Pick a single colony and mix it in 10μL sterile water, then use 0.5 μL of the mixture as the template for PCR. Colony PCR system:
2×Taq PCR mix | 10 μL |
Primer 1(10μM) | 0.5 μL |
Primer 2(10μM) | 0.5 μL |
Bacterial liquid | 0.5 μL |
ddH2O | Make up to 20 μL |
Total | 20 μL |
Colony PCR program: Preheat the PCR reaction system to 94°C for 5 minutes to fully denature the template DNA. Then, perform 30 cycles of amplification, each including 30 seconds of denaturation at 94°C, 30 seconds of annealing at 55°C, and 30 seconds of extension at 72°C. Store at 10°C until the PCR product is collected.
3.2.6. Transformation of Bacillus subtilis
Bacillus subtilis was inoculated in 3 mL LB medium and cultured overnight to obtain sufficient bacterial biomass. Then, 2.6 mL of the overnight culture was transferred to a 40 mL LB medium containing 0.5M sorbitol and incubated at 37°C, 200 rpm, until OD600 reached 0.85–0.95. The bacterial suspension was then chilled on ice for 10 minutes, centrifuged at 5000 g, 4°C for 5 minutes, and resuspended in a pre-cooled electroporation medium. After four washing steps, the cells were resuspended in 1 mL electroporation buffer and aliquoted for electroporation experiments.
3.2.7. Spore Induction
Spores were induced in the DSM medium by inoculating the strain and incubating at 37°C for 48 hours. After centrifugation, the pellet was resuspended in PBS and treated with lysozyme to lyse vegetative cells. The spore suspension was washed twice with NaCl and KCl solutions and further purified by heating at 70°C for 1 hour to inactivate residual vegetative cells.
3.2.8. Extraction of Spore Coat Proteins
Spores were mixed with buffer containing NaCl, NaOH, SDS, and dithiothreitol (DTT), then incubated at 70°C for 1 hour. After centrifugation, the supernatant was discarded, and the pellet was resuspended in PBS. The protein concentration was measured using a BCA protein assay kit.
3.2.9. Protein Blotting
To analyze the expression of spore coat proteins, the proteins were separated by 12% SDS-PAGE and transferred onto PVDF membranes. The membranes were blocked with 3% BSA for 2 hours and washed with TBST. After overnight incubation with anti-polyhistidine monoclonal antibody at 4°C, the membranes were washed again and incubated with HRP-conjugated goat anti-mouse IgG. The signal was detected using an ECL kit and visualized with a gel imaging system.
3.2.10.Immunofluorescence Analysis
Spore cultures (500 μL) were washed with PBS and blocked with 3% BSA for 30 minutes. After washing with TBST, the samples were incubated with anti-polyhistidine monoclonal antibody at 4°C overnight. The samples were then incubated with FITC-conjugated anti-mouse IgG for 2 hours and analyzed using a confocal laser scanning microscope (Olympus, FV4000) with LAS AF Lite software.
3.2.11.Enzyme Activity Analysis
Enzyme activity of recombinant spores was measured by detecting the UV absorption peaks at 260 nm for C12O and 375 nm for C23O. One unit of enzyme activity was defined as the amount of spore required to oxidize 1 μmol of catechol per minute. The activity and stability of recombinant spores were tested across a pH range of 3–11 and temperatures from 0°C to 80°C. Tolerance to extreme conditions, including high temperature (60°C), low temperature (-20°C), organic solvents (methanol, diethyl ether), proteases (trypsin, proteinase), and pH extremes (pH 4 and pH 10), was also evaluated.
4. Research results and analysis
4.1. Construction of Recombinant Plasmids
Based on literature research, two catechol-degrading enzymes, C12O and C23O, were selected for study. Their sequences are WP_332871064.1 and KF261118, respectively. Primers with flexible linkers [Gly-Gly-Gly-Gly-Ser] and a 6×His tag were designed for gene cloning and vector construction (Figure 1).
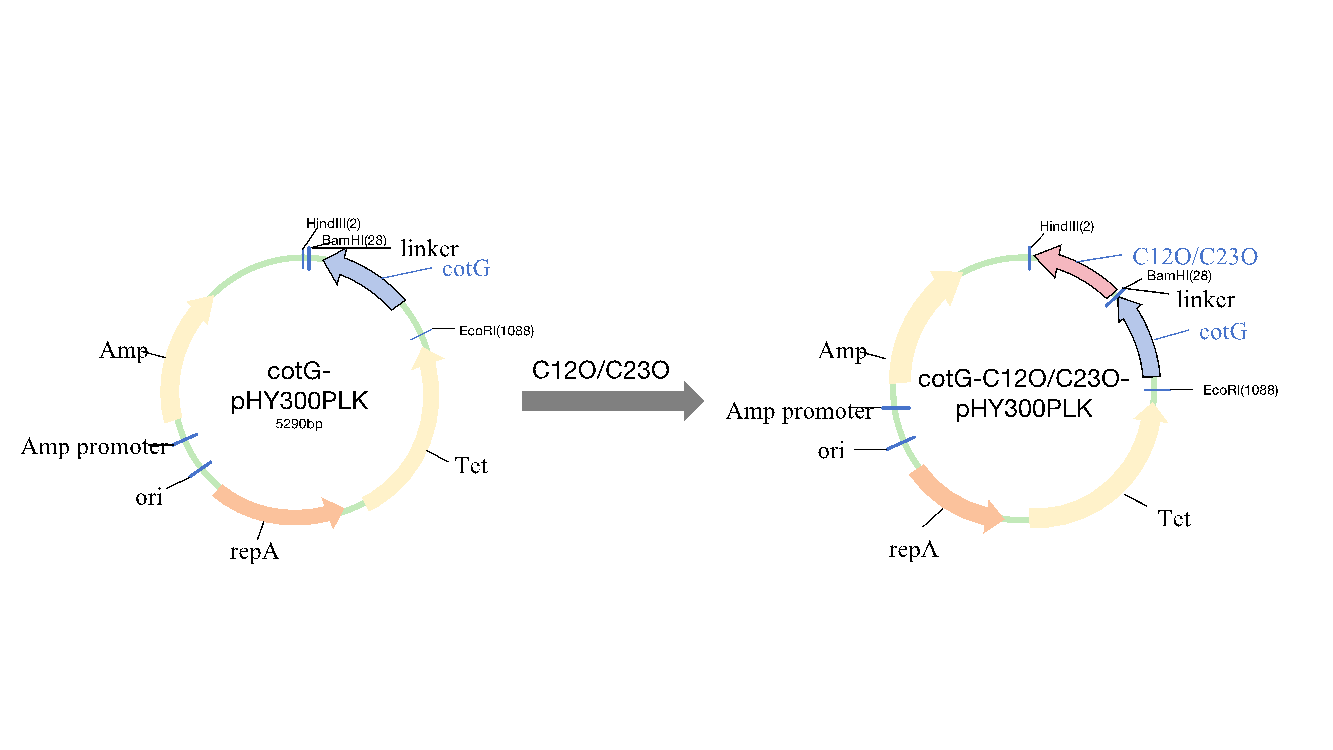
Figure 1: Schematic diagram of recombinant plasmid construction
Using total DNA from Bacillus subtilis and synthetic C12O and C23O genes, the cotG anchoring protein and the catechol dioxygenases C12O and C23O were PCR-amplified. The amplification results showed single bands for all three genes (Figure 2). The cloned fragments were sequenced and confirmed to match the published GenBank sequences. The shuttle vector pHY300plk, used for genetic manipulation in E. coli and Bacillus subtilis, was digested, purified, and ligated with the gene fragments, then transformed into E. coli DH5α. Positive clones were selected and identified by colony PCR (Figure 3). After sequencing, the plasmids were confirmed to contain the correct gene sequence and transformed into Bacillus subtilis DB104. The transformed colonies were confirmed by colony PCR (Figure 4). The resulting strains were named FZ00, FZ01, and FZ02, where FZ00 is the control (pHY300PLK-cotG), FZ01 expresses C12O, and FZ02 expresses C23O
.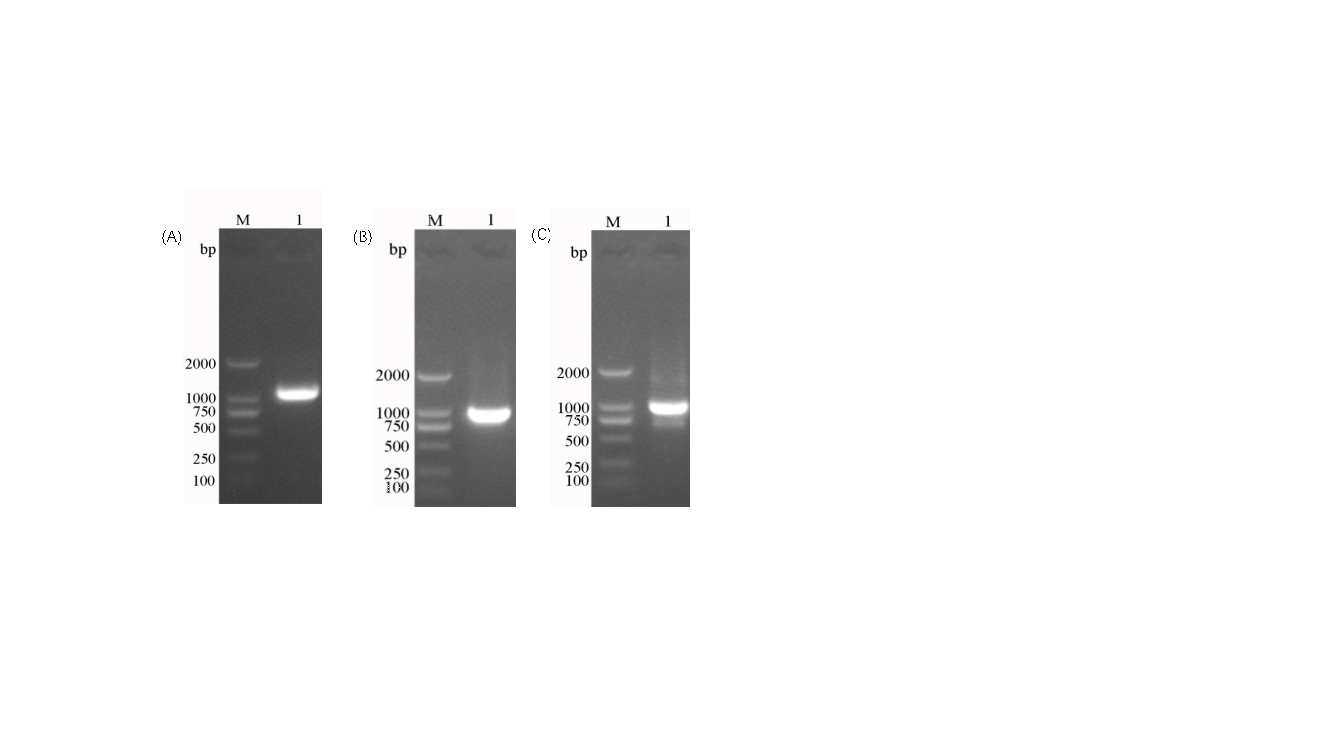
Figure 2: Cloning of anchoring protein and catechol dioxygenase genes. A: cotG; B: C12O; C: C23O.
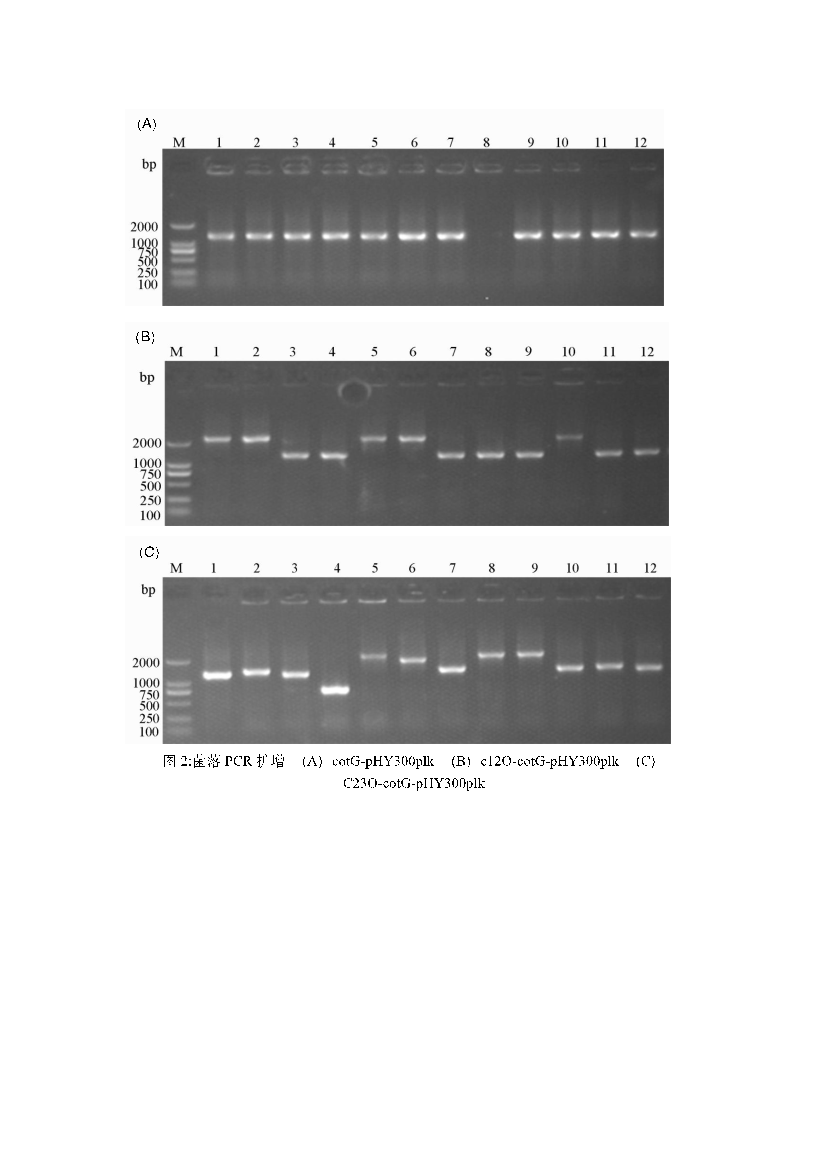
Figure 3: PCR amplification of E. coli colonies. A: pHY300PLK-cotg; B: pHY300PLK-cotg-C12O-6×his; C: pHY300PLK-cotg-C23O-6×his.
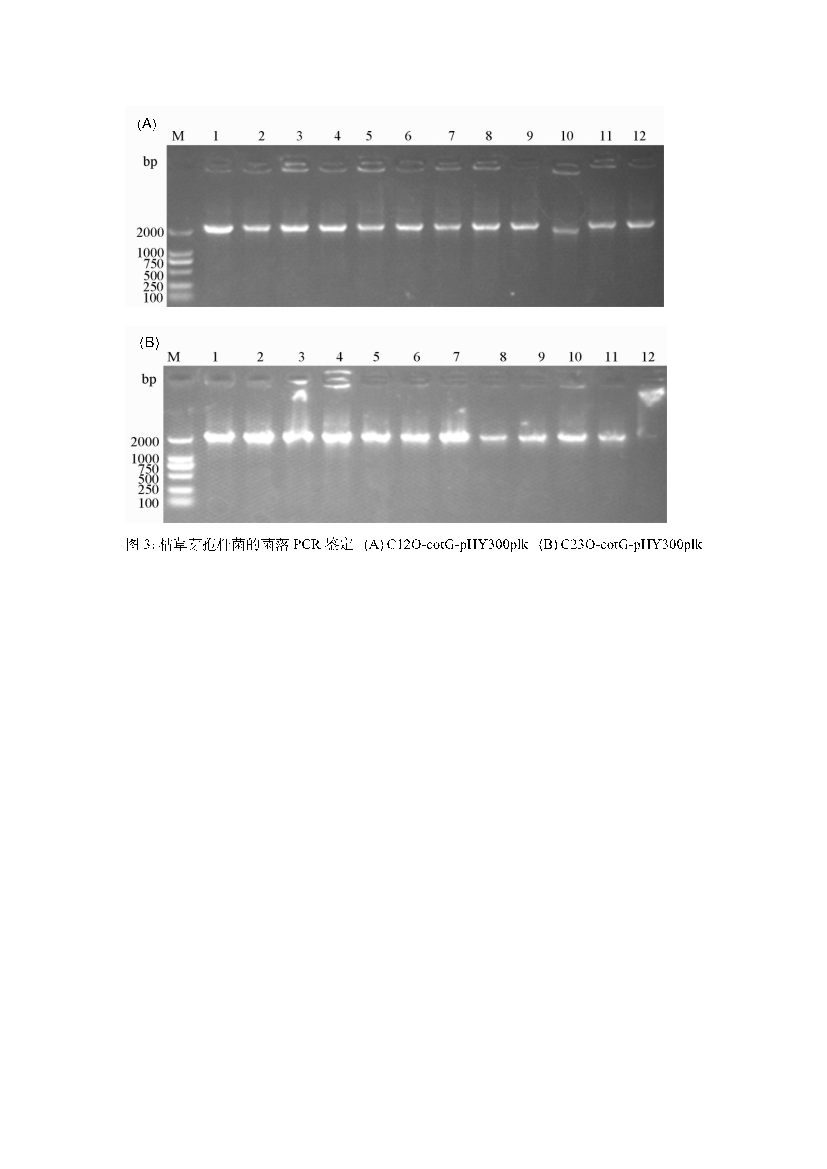
Figure 4: PCR amplification of Bacillus subtilis colonies. A: pHY300PLK-cotg-C12O-6×his; B: pHY300PLK-cotg-C23O-6×his.
4.2. Expression of C12O and C23O in Recombinant Spores
To verify the expression of C12O and C23O on the spore surface, Western blot analysis was performed. Spore coat proteins were extracted and detected using anti-His tag antibodies. Specific bands of approximately 59 kDa were observed, confirming that C12O and C23O were successfully displayed on the spore surface. No signal was detected from the FZ00 control spores, confirming that the observed signals were from the recombinant proteins (Figure 5).
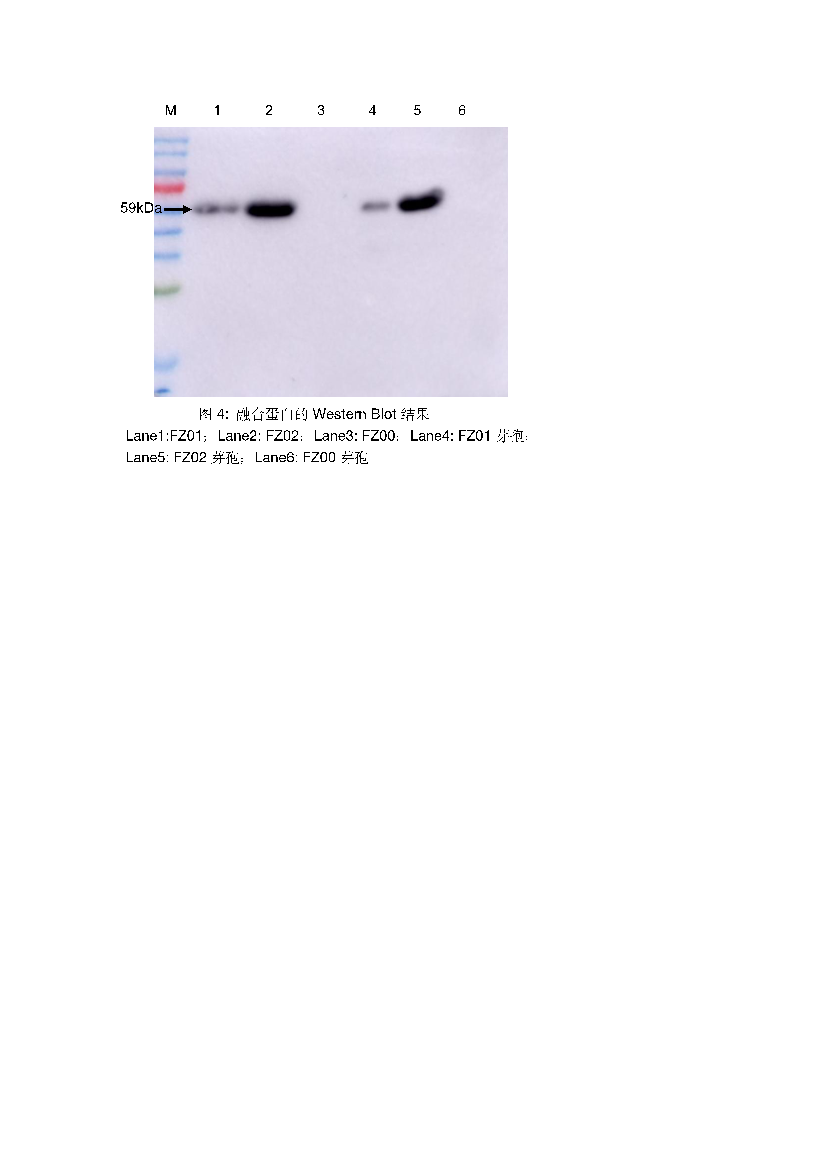
Figure 5: Western Blot results of fusion protein. Lane 1: FZ01; Lane 2: FZ02; Lane 3: FZ00; Lane 4: FZ01 spores; Lane 5: FZ02 spores; Lane 6: FZ00 spores.
4.3. Surface Display of C12O and C23O on Spores
Immunofluorescence microscopy was used to analyze the expression and localization of C12O and C23O on the surface of recombinant Bacillus subtilis spores (FZ01 and FZ02). In the fluorescence images of FZ01 spores (second column, Figure 6), green fluorescence signals were observed, indicating a successful surface display of C12O. The signal was detected using an anti-His tag antibody, and the green fluorescence represented the binding of the antibody to the displayed protein. Similar fluorescence signals were observed in FZ02 spores, indicating a successful display of C23O. Overlay images (Merge) showed clear co-localization of green fluorescence with the spore contours, confirming the correct localization of the proteins on the spore surface. No fluorescence signal was detected in FZ00 spores, confirming the absence of recombinant protein expression in the control strain.

Figure 6: Immunofluorescence microscopy of recombinant spores. Phase contrast: bright field microscopic images of all spores; Fluorescence: green fluorescence indicates that the 6×His-tagged antibody specifically binds to pHY300PLK-cotg-C12O/C23O-6his-tagged protein; Merge: bright field and fluorescence superposition images.
4.4. Enzymatic Properties of Recombinant Spores
4.4.1. Catechol Degradation Activity
Compared to FZ00, which does not express C12O, the recombinant FZ01 spores showed significantly higher catechol degradation activity. FZ01 had an activity of 4.517 U/mg, 10.5 times higher than FZ00 (Figure 7A). Similarly, FZ02, which expresses C23O, showed 2.732 U/mg activity, 14.1 times higher than FZ00 (Figure 7B). These results confirm that C12O and C23O were successfully expressed on the spore surface and possess catechol degradation activity.
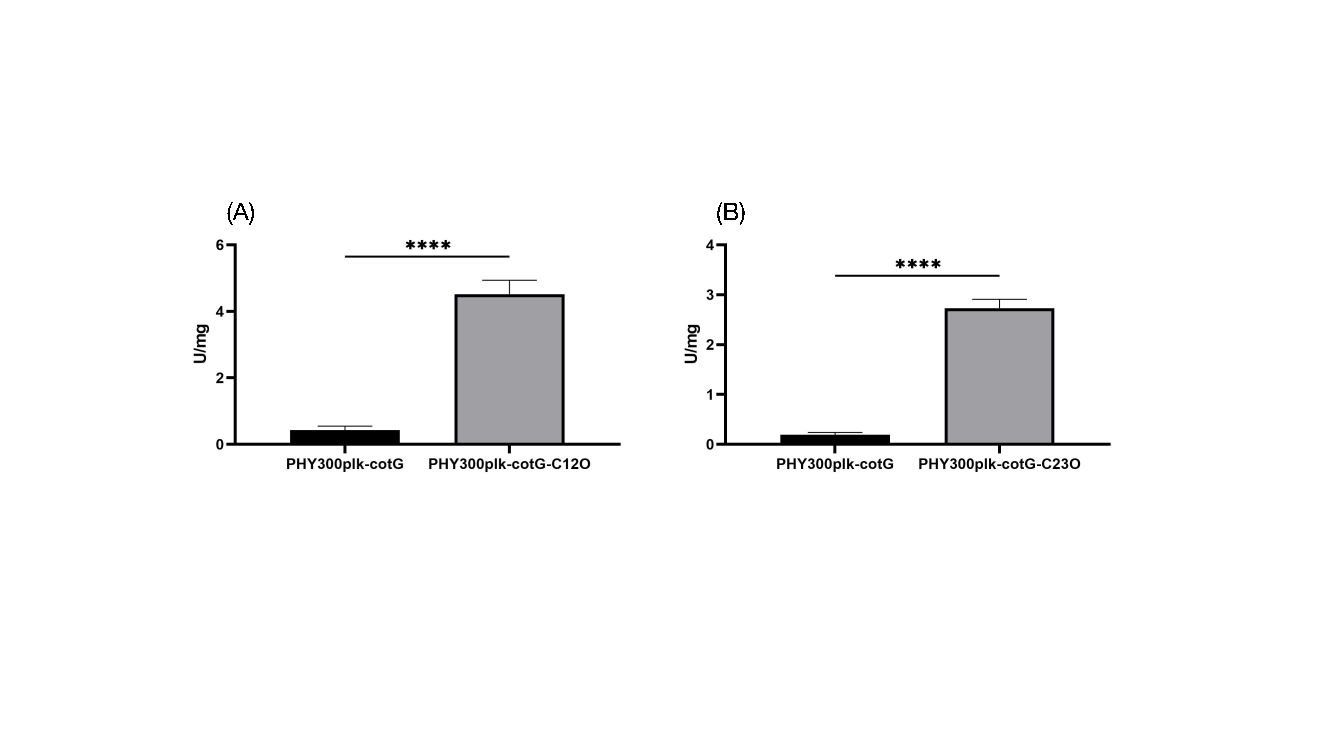
Figure 7: Catechol degradation activity of recombinant spores. A: FZ01 spores; B: FZ02 spores.
4.4.2. Activity and Stability of Recombinant Spores under Different pH and Temperature Conditions
To assess the practical application of recombinant spores, their activity and stability were tested at pH values ranging from 3 to 11 and temperatures from 0°C to 80°C (Figures 8 and 9). FZ01 spores had optimal pH between 7 and 8, maintaining over 60% activity across pH 5–10. At extreme pH values (pH 3 and 11), FZ01 retained 18.4% and 38.2% activity, respectively. The optimal temperature for FZ01 was 30°C, with 24.3% activity at 0°C and 16.5% at 80°C. FZ02 spores had a similar pH profile, with optimal activity at pH 7–8 and stable activity at extreme pH conditions (10.6% at pH 3 and 28.4% at pH 11). Their optimal temperature was 30°C–40°C, maintaining 14.4% activity at 0°C and 8.5% at 80°C.
Both FZ01 and FZ02 spores demonstrated strong stability under various pH and temperature conditions. After 1 hour of treatment, FZ01 maintained 91.7% activity at 0°C and 20.8% at 80°C. After 24 hours, FZ01 retained 82.7% activity at an optimal pH and 89.9% at an optimal temperature. FZ02 spores showed similar stability, with 96.3% activity at optimal pH after 1 hour, and 98.7% activity at optimal temperature.
These findings demonstrate that C12O and C23O expressed on the spore surface exhibit broad functionality under various pH and temperature conditions, making them promising for industrial applications.
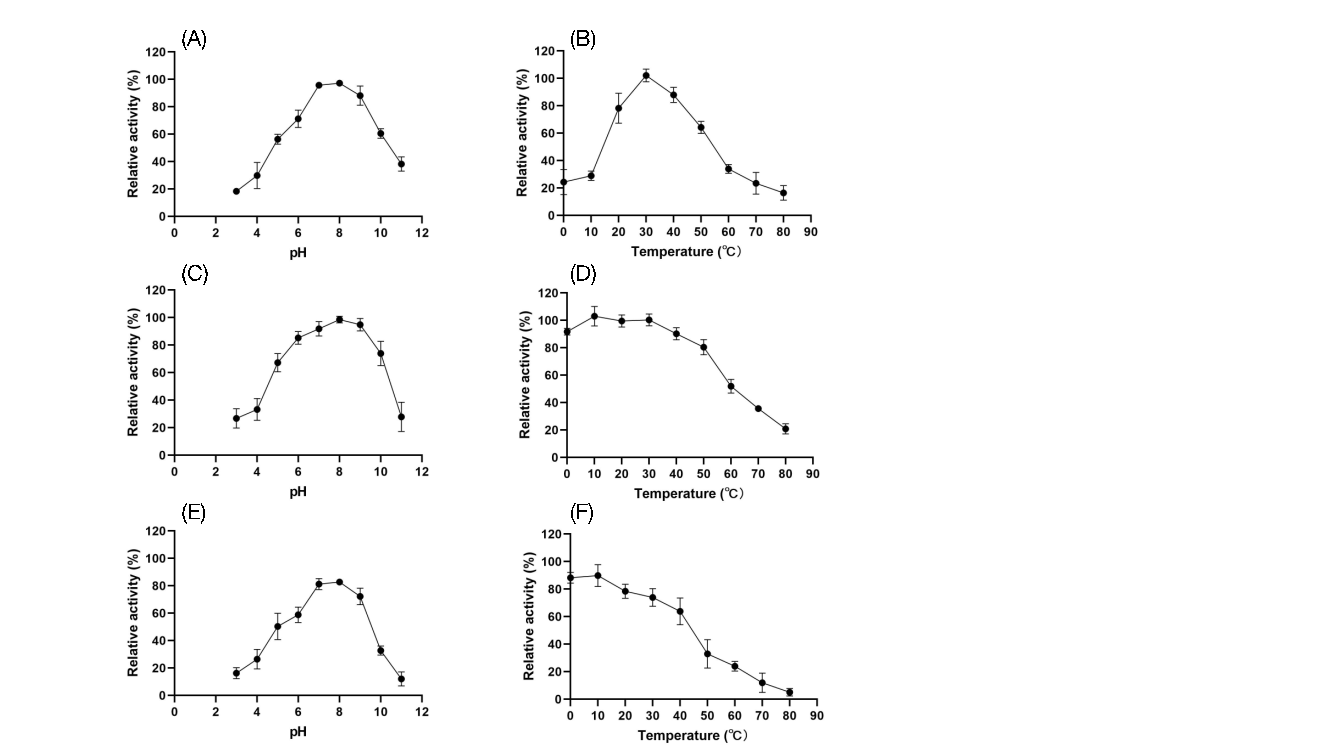
Figure 8: Activity and stability of FZ01 spores under different pH and temperature conditions. A: Activity under different pH conditions; B: Activity under different temperature conditions; C: Stability after 1 hour of treatment under different pH conditions; D: Stability after 1 hour of treatment under different temperature conditions; E: Stability after 1 day of treatment under different pH conditions; F: Stability after 1 day of treatment under different temperature conditions.
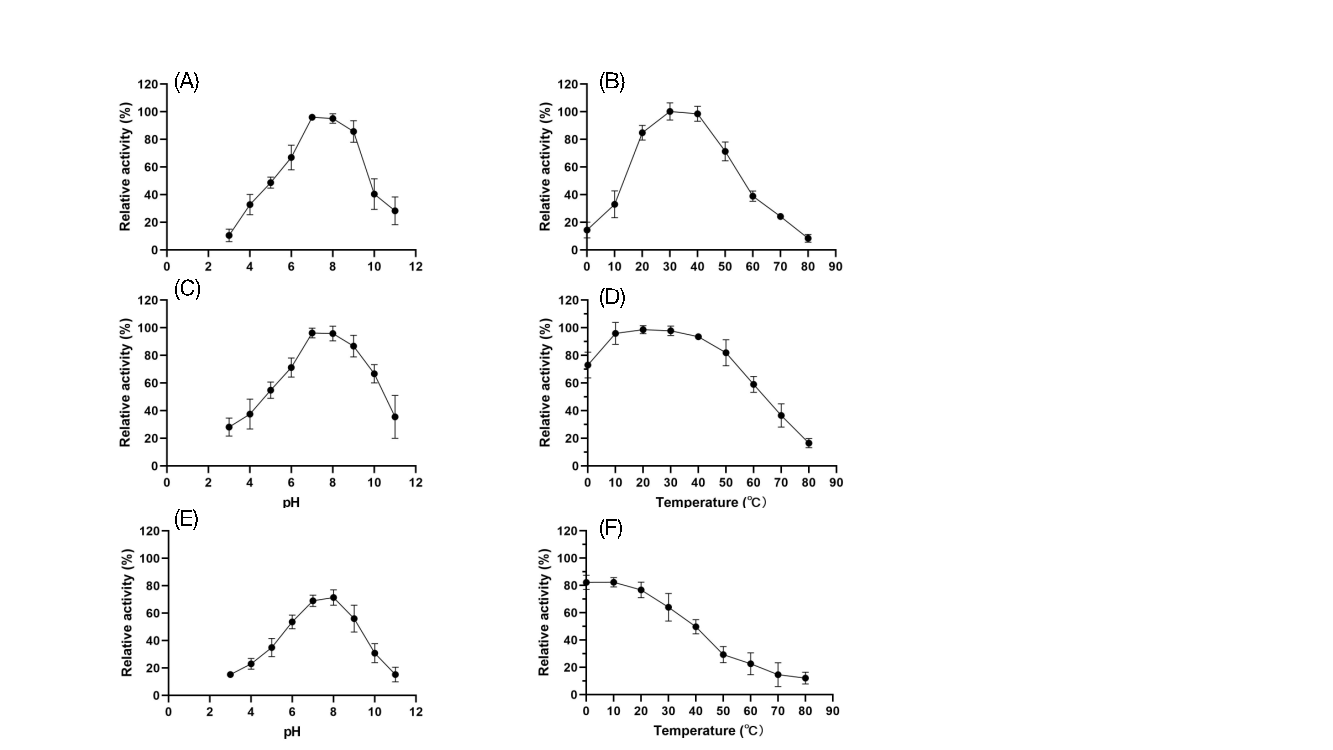
Figure 9: Activity and stability of FZ02 spores under different pH and temperature conditions. A: Activity under different pH conditions; B: Activity under different temperature conditions; C: Stability after 1 hour of treatment under different pH conditions; D: Stability after 1 hour of treatment under different temperature conditions; E: Stability after 1 day of treatment under different pH conditions; F: Stability after 1 day of treatment under different temperature conditions.
4.4.3. Tolerance of Recombinant Spores under Extreme Conditions
After 1 hour of treatment under various extreme conditions, FZ01 spores showed a relative activity range of 10%-95%, with methanol-treated spores exhibiting the highest activity and 60°C-treated spores the lowest. Other conditions resulted in 12%-32% activity. FZ02 spores displayed a similar range of 5%-95%, with methanol-treated spores maintaining nearly full activity, while the lowest activity was observed in spores treated at pH 4. Other conditions yielded 20%-35% activity. These results suggest that both FZ01 and FZ02 spores show strong tolerance to organic solvents, particularly methanol, indicating potential for further testing in environmental applications.
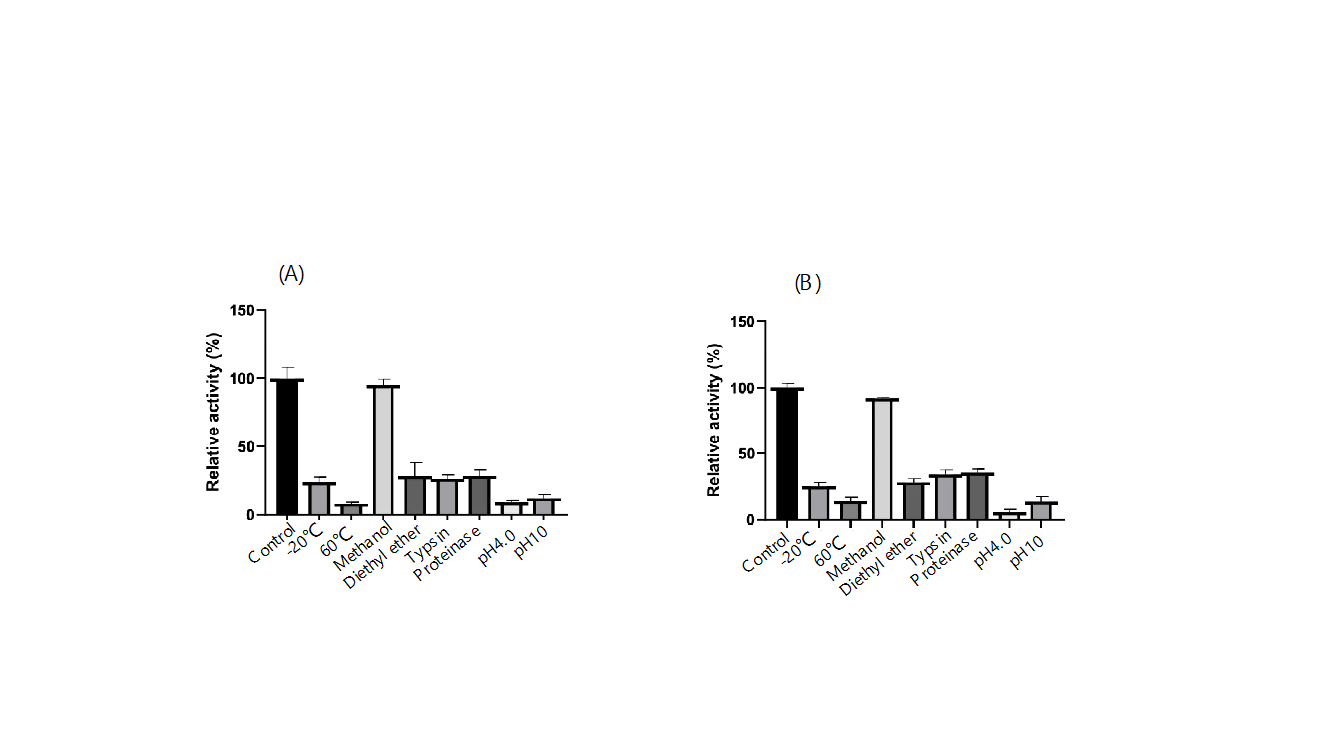
Figure 10: Tolerance of recombinant spores under different extreme conditions. (A) FZ01; (B) FZ02.
5. Discussion
5.1. Results Analysis
In this study, the Bacillus subtilis spore surface display system was successfully applied to the biodegradation of catechol, demonstrating its effectiveness in pollution control. By constructing and transforming recombinant vectors with C12O and C23O genes, these enzymes were displayed on the Bacillus subtilis spore surface. Western blot and fluorescence microscopy confirmed the successful expression of C12O and C23O with the aid of the cotG anchoring protein. Enzyme activity analysis showed that both enzymes maintained catechol-degrading activity across a range of pH and temperature conditions and exhibited good stability in organic solvents like methanol, suggesting their potential for practical applications.
When comparing the enzymatic properties of FZ01 and FZ02 spores to previously reported C12O and C23O, we found advantages for the spore surface display. For instance, although FZ01 spores exhibited lower enzyme activity than purified C12O protein, the activity per spore weight could be higher due to the enzyme’s immobilization on the spore surface. Similarly, although FZ02 spores had lower activity than purified C23O, this might be due to the tetrameric form of C23O being more spatially constrained on the spore surface. Overall, displaying C12O and C23O on spores not only removes the purification step, reducing costs, but in some cases, it can offer better degradation results, especially for C12O. Additionally, exposure of spores to 70°C for 1 hour to remove remaining vegetative cells might affect enzyme activity on the spore surface, suggesting that eliminating this step could improve degradation in practical applications.
Furthermore, comparisons with purified proteins revealed that FZ01 and FZ02 spores retained higher activity across a broader pH and temperature range than purified enzymes. Notably, the spores maintained over 10% activity in the extreme pH range of 3-11, outperforming purified proteins. The spores also retained significant activity at 80°C, indicating better heat tolerance compared to the purified enzymes.
Lastly, the FZ01 and FZ02 spores exhibited strong resistance to organic solvents, especially methanol, suggesting that surface-displayed C12O and C23O could have significant potential in environments with organic solvents. Overall, the study shows that displaying enzymes on spores significantly enhances their tolerance to extreme pH, temperature, and organic solvents.
5.2. Innovations of this Study
The core innovation of this project lies in applying the Bacillus subtilis spore surface display system to the biodegradation of catechol, a first in the biotechnology field. Catechol, a widely used aromatic compound in industrial production, is difficult to degrade, posing significant environmental challenges. Traditional physical and chemical methods are costly and may cause secondary pollution. In contrast, this study offers a biological degradation method with low cost and no pollution, showcasing its unique advantages.
By genetically engineering Bacillus subtilis spores to display the catechol-degrading enzymes C12O and C23O, this approach simplifies traditional biodegradation methods by eliminating the need for protein purification, reducing costs, and leveraging the natural resilience of spores. The enzymes displayed on spores maintain high activity across a wide pH and temperature range and even in harsh chemical environments, greatly expanding the potential applications of biodegradation technologies.
Moreover, environmental adaptability tests of the displayed C12O and C23O enzymes, including activity and tolerance assessments in different pH, temperature, and organic solvent conditions, provide valuable data for optimizing biodegradation strategies in real-world environmental remediation.
5.3. Current Challenges and Future Directions
Although this study successfully demonstrated the Bacillus subtilis spore surface display system for catechol degradation, several challenges remain. First, enzyme activity on spores was lower than that of purified C12O and C23O, likely due to spatial constraints or microenvironmental factors on the spore surface. Second, the enzymes displayed via cotG exhibited reduced tolerance to proteases and organic solvents (except methanol), possibly due to their tetrameric forms. Future research could explore alternative anchoring proteins, such as cotB and cotC, which are commonly used in spore surface display systems, to balance enzyme activity and stability.
Additionally, methods for large-scale production and purification of recombinant spores need to be developed for industrial applications. While laboratory results are promising, real-world environmental complexity may affect the spores' degradation efficiency and stability, necessitating further testing in diverse environmental conditions. Moreover, although biological degradation offers a cost advantage, detailed economic analyses comparing this method with traditional approaches are still needed.
Future research should focus on improving enzyme efficiency by optimizing display conditions, scaling up production processes, and conducting cost-benefit analyses. Safety assessments, including potential environmental and human health impacts, are essential to ensure the safety of the application. Finally, developing regulatory frameworks to comply with environmental protection and biosafety standards will be crucial for the widespread adoption of this technology.
Table 5: pH and temperature ranges and tolerance ranges of C12O reported in the literature
Species | pH range of activity | pH range of stability | Temperature range of activity | Temperature range of stability | |
Pseudomonas aeruginosa | 5(0)-10(65) | 5(0)-10(0) | 20(30)-70(0) | 60(0)* | [16] |
Sphingomonas xenophaga | 4.6(0)-10.7(50) | -- | 20(48)-70(82) | -- | [17] |
Acinetobacter sp. | 3(42)-11(38) | 3(0)-10(0) | 25(35)-50(17) | 55(0)* | [18] |
Cupriavidus campinensis | 4(25)-12(0) | 5(0)-11(0) | 20(86)-60(5) | 50(0)* | [19] |
Oceanimonas marisflavi | 2(5)-11(25) | 2(8)-11(30) | 20(55)-70(0) | 70(0)* | [20] |
Gordonia polyisoprenivorans | 4(5)-9(30) | -- | 5(5)-50(0) | -- | [1] |
*Indicates that only the upper-temperature tolerance limit was measured. The data in brackets are the relative activity or residual activity at that temperature or pH. The bold data are the data of the purified protein of C12O used in this study.
Table 6: pH and temperature ranges and tolerance ranges of C23O reported in the literature
Species | pH range of activity | pH range of stability | Temperature range of activity | Temperature range of stability | |
Stenotrophomonas maltophilia | 4.7(0)-9(23) | -- | 6(0)-61(0) | -- | [21] |
Bacillus ligniniphilus | 6(0)-8.5(7) | -- | 25(90)-45(50) | 56(9.6)* | [22] |
Halophilic bacterial consortium | -- | -- | 0(19)-80(3) | 4(50)-50(0) | [11] |
Halophilic bacterial consortium | -- | -- | 0(0)-80(41) | 4(50)-50(0) | [11] |
Gordonia polyisoprenivorans | 4(30)-9(65) | -- | 5(13)-50(37) | -- | [1] |
Stenotrophomonas maltophilia | 2.2(0)-10(0) | -- | 5(57)-55(0) | -- | [12] |
*Indicates that only the upper-temperature tolerance limit was measured. The data in brackets are the relative activity or residual activity at that temperature or pH. The bold data are the data of the purified protein of C12O used in this study.
6. Conclusion
This study successfully applied the Bacillus subtilis spore surface display system to the biodegradation of catechol, providing an innovative biotechnological strategy for environmental pollution control. The recombinant spores FZ01 and FZ02 successfully expressed C12O and C23O on their surfaces, and their catechol-degrading activity and environmental tolerance were comprehensively evaluated.
The results indicate that the surface-displayed enzymes maintained good activity and stability across a broad pH (3-11) and temperature (0-80°C) range, especially in the presence of organic solvents like methanol, highlighting their potential for real-world environmental remediation. Compared to purified C12O and C23O, the spore-bound enzymes not only eliminated purification steps but also demonstrated superior degradation performance in some conditions, particularly for C12O.
However, several challenges remain, including the need to improve enzyme efficiency, develop scalable production methods, and conduct a detailed economic analysis. Future research should address these issues and further evaluate the safety, feasibility, and regulatory compliance of this biotechnological approach for environmental applications.
References
[1]. Silva, A.S., et al., Enzymatic activity of catechol 1, 2-dioxygenase and catechol 2, 3-dioxygenase produced by Gordonia polyisoprenivorans. Quimica Nova, 2012. 35: p. 1587-1592.
[2]. Aravind, M.K., et al., Bioengineered Graphene Oxide Microcomposites Containing Metabolically Versatile Paracoccus sp. MKU1 for Enhanced Catechol Degradation. ACS Omega, 2020. 5(27): p. 16752-16761.
[3]. Topping, D.C., et al., Hydroquinone: acute and subchronic toxicity studies with emphasis on neurobehavioral and nephrotoxic effects. Food and chemical toxicology, 2007. 45(1): p. 70-78.
[4]. Zhou, Y., et al., Mesoporous carbon nitride based biosensor for highly sensitive and selective analysis of phenol and catechol in compost bioremediation. Biosensors and Bioelectronics, 2014. 61: p. 519-525.
[5]. Kubesch, K., et al., Degradation of catechol by ionizing radiation, ozone and the combined process ozone-electron-beam. Radiation Physics and Chemistry, 2005. 72(4): p. 447-453.
[6]. Ledesma, E.B., et al., An experimental study on the thermal decomposition of catechol. Proceedings of the Combustion Institute, 2002. 29(2): p. 2299-2306.
[7]. Xiao, J., et al., Enhancement of Fenton degradation by catechol in a wide initial pH range. Separation and Purification Technology, 2016. 169: p. 202-209.
[8]. Zhang, H., X. Bo, and L. Guo, Electrochemical preparation of porous graphene and its electrochemical application in the simultaneous determination of hydroquinone, catechol, and resorcinol. Sensors and Actuators B: Chemical, 2015. 220: p. 919-926.
[9]. El Azhari, N., et al., Molecular analysis of the catechol-degrading bacterial community in a coal wasteland heavily contaminated with PAHs. J Hazard Mater, 2010. 177(1-3): p. 593-601.
[10]. Vesely, M., et al., Analysis of catRABC operon for catechol degradation from phenol-degrading Rhodococcus erythropolis. Appl Microbiol Biotechnol, 2007. 76(1): p. 159-68.
[11]. Guo, G., et al., Isolation and characterization of two novel halotolerant Catechol 2, 3-dioxygenases from a halophilic bacterial consortium. Sci Rep, 2015. 5: p. 17603.
[12]. Wojcieszyńska, D., et al., Properties of catechol 2,3-dioxygenase from crude extract of Stenotrophomonas maltophilia strain KB2 immobilized in calcium alginate hydrogels. Biochemical Engineering Journal, 2012. 66: p. 1-7.
[13]. Bartels, J., et al., Sporobeads: The Utilization of the Bacillus subtilis Endospore Crust as a Protein Display Platform. ACS Synth Biol, 2018. 7(2): p. 452-461.
[14]. Wang, H., Y. Wang, and R. Yang, Recent progress in Bacillus subtilis spore-surface display: concept, progress, and future. Appl Microbiol Biotechnol, 2017. 101(3): p. 933-949.
[15]. Tavassoli, S., et al., Investigation of spore coat display of Bacillus subtilis beta-galactosidase for developing of whole cell biocatalyst. Arch Microbiol, 2013. 195(3): p. 197-202.
[16]. Wang, C.-L., S.-L. You, and S.-L. Wang, Purification and characterization of a novel catechol 1,2-dioxygenase from Pseudomonas aeruginosa with benzoic acid as a carbon source. Process Biochemistry, 2006. 41(7): p. 1594-1601.
[17]. Gou, M., et al., Characterization of catechol 1,2-dioxygenase from cell extracts of Sphingomonas xenophaga QYY. Science in China Series B: Chemistry, 2009. 52(5): p. 615-620.
[18]. Lin, J. and R.N. Milase, Purification and Characterization of Catechol 1,2-Dioxygenase from Acinetobacter sp. Y64 Strain and Escherichia coli Transformants. Protein J, 2015. 34(6): p. 421-33.
[19]. Han, L., S. Chen, and J. Zhou, Expression and cloning of catA encoding a catechol 1,2-dioxygenase from the 2,4-D-degrading strain Cupriavidus campinensis BJ71. Prep Biochem Biotechnol, 2020. 50(5): p. 486-493.
[20]. Li, J., et al., Expression and characterization of catechol 1,2-dioxygenase from Oceanimonas marisflavi 102-Na3. Protein Expr Purif, 2021. 188: p. 105964.
[21]. Guzik, U., et al., High activity catechol 1,2-dioxygenase from Stenotrophomonas maltophilia strain KB2 as a useful tool in cis,cis-muconic acid production. Antonie Van Leeuwenhoek, 2013. 103(6): p. 1297-307.
[22]. Adewale, P., et al., A novel Bacillus ligniniphilus catechol 2,3-dioxygenase shows unique substrate preference and metal requirement. Sci Rep, 2021. 11(1): p. 23982.
Cite this article
Fu,Z. (2025). Exploring the Application of Bacillus Subtilis Spore Surface Display System in the Field of Catechol Degradation. Theoretical and Natural Science,99,1-15.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 5th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Silva, A.S., et al., Enzymatic activity of catechol 1, 2-dioxygenase and catechol 2, 3-dioxygenase produced by Gordonia polyisoprenivorans. Quimica Nova, 2012. 35: p. 1587-1592.
[2]. Aravind, M.K., et al., Bioengineered Graphene Oxide Microcomposites Containing Metabolically Versatile Paracoccus sp. MKU1 for Enhanced Catechol Degradation. ACS Omega, 2020. 5(27): p. 16752-16761.
[3]. Topping, D.C., et al., Hydroquinone: acute and subchronic toxicity studies with emphasis on neurobehavioral and nephrotoxic effects. Food and chemical toxicology, 2007. 45(1): p. 70-78.
[4]. Zhou, Y., et al., Mesoporous carbon nitride based biosensor for highly sensitive and selective analysis of phenol and catechol in compost bioremediation. Biosensors and Bioelectronics, 2014. 61: p. 519-525.
[5]. Kubesch, K., et al., Degradation of catechol by ionizing radiation, ozone and the combined process ozone-electron-beam. Radiation Physics and Chemistry, 2005. 72(4): p. 447-453.
[6]. Ledesma, E.B., et al., An experimental study on the thermal decomposition of catechol. Proceedings of the Combustion Institute, 2002. 29(2): p. 2299-2306.
[7]. Xiao, J., et al., Enhancement of Fenton degradation by catechol in a wide initial pH range. Separation and Purification Technology, 2016. 169: p. 202-209.
[8]. Zhang, H., X. Bo, and L. Guo, Electrochemical preparation of porous graphene and its electrochemical application in the simultaneous determination of hydroquinone, catechol, and resorcinol. Sensors and Actuators B: Chemical, 2015. 220: p. 919-926.
[9]. El Azhari, N., et al., Molecular analysis of the catechol-degrading bacterial community in a coal wasteland heavily contaminated with PAHs. J Hazard Mater, 2010. 177(1-3): p. 593-601.
[10]. Vesely, M., et al., Analysis of catRABC operon for catechol degradation from phenol-degrading Rhodococcus erythropolis. Appl Microbiol Biotechnol, 2007. 76(1): p. 159-68.
[11]. Guo, G., et al., Isolation and characterization of two novel halotolerant Catechol 2, 3-dioxygenases from a halophilic bacterial consortium. Sci Rep, 2015. 5: p. 17603.
[12]. Wojcieszyńska, D., et al., Properties of catechol 2,3-dioxygenase from crude extract of Stenotrophomonas maltophilia strain KB2 immobilized in calcium alginate hydrogels. Biochemical Engineering Journal, 2012. 66: p. 1-7.
[13]. Bartels, J., et al., Sporobeads: The Utilization of the Bacillus subtilis Endospore Crust as a Protein Display Platform. ACS Synth Biol, 2018. 7(2): p. 452-461.
[14]. Wang, H., Y. Wang, and R. Yang, Recent progress in Bacillus subtilis spore-surface display: concept, progress, and future. Appl Microbiol Biotechnol, 2017. 101(3): p. 933-949.
[15]. Tavassoli, S., et al., Investigation of spore coat display of Bacillus subtilis beta-galactosidase for developing of whole cell biocatalyst. Arch Microbiol, 2013. 195(3): p. 197-202.
[16]. Wang, C.-L., S.-L. You, and S.-L. Wang, Purification and characterization of a novel catechol 1,2-dioxygenase from Pseudomonas aeruginosa with benzoic acid as a carbon source. Process Biochemistry, 2006. 41(7): p. 1594-1601.
[17]. Gou, M., et al., Characterization of catechol 1,2-dioxygenase from cell extracts of Sphingomonas xenophaga QYY. Science in China Series B: Chemistry, 2009. 52(5): p. 615-620.
[18]. Lin, J. and R.N. Milase, Purification and Characterization of Catechol 1,2-Dioxygenase from Acinetobacter sp. Y64 Strain and Escherichia coli Transformants. Protein J, 2015. 34(6): p. 421-33.
[19]. Han, L., S. Chen, and J. Zhou, Expression and cloning of catA encoding a catechol 1,2-dioxygenase from the 2,4-D-degrading strain Cupriavidus campinensis BJ71. Prep Biochem Biotechnol, 2020. 50(5): p. 486-493.
[20]. Li, J., et al., Expression and characterization of catechol 1,2-dioxygenase from Oceanimonas marisflavi 102-Na3. Protein Expr Purif, 2021. 188: p. 105964.
[21]. Guzik, U., et al., High activity catechol 1,2-dioxygenase from Stenotrophomonas maltophilia strain KB2 as a useful tool in cis,cis-muconic acid production. Antonie Van Leeuwenhoek, 2013. 103(6): p. 1297-307.
[22]. Adewale, P., et al., A novel Bacillus ligniniphilus catechol 2,3-dioxygenase shows unique substrate preference and metal requirement. Sci Rep, 2021. 11(1): p. 23982.