







1. Introduction
Breast cancer (BC) remains a significant global health concern, which represents the most frequently diagnosed malignancy in women. It accounts for 10% of all cancer incidences in women and is the second leading cause of cancer-related deaths in the United States [1]. The growing number of cases can be attributed to a complex interplay of genetic predispositions and non-genetic risk factors such as age, reproductive history, hormonal exposure and environmental influences [2,3].
In modern molecular pathology, high-throughput biomarker screening has offered a convincing explanation for BC heterogeneity and key biomarkers are used for molecular stratification of BC [4], including hormone receptor (HR) positive breast cancer, HER2-positive breast cancer and triple-negative breast cancer (TNBC) [5]. HR positive breast cancers, characterized by the expression of estrogen receptor (ER) and/or progesterone receptor (PR), represent the most prevalent subtype of BC and are associated with both the highest incidence and recurrence rates among BCClinically, HR+ BCs are typically well-differentiated, exhibit lower aggressiveness, and are associated with a more favorable prognosis compared to HR- BCs [6]. Consequently, endocrine therapy is suggested for most patients with HR+ BCs. Tamoxifen is utilized in both premenopausal and postmenopausal women, whereas aromatase inhibitors (Ais), including anstrozole, letrozole and exemestane, are exclusively administered in postmenopausal women [7]. AIs are generally favored over tamoxifen for adjuvant therapy, though a sequential regimen combining tamoxifen followed by Ais may also be employed to optimize therapeutic outcomes [7].
HER2+ BC represents approximately 20% of BC, which is defined by the abnormal amplification of the ERBB2/neu oncogene [7,8] and associated with high levels of genetic instability and irregularities in cell morphology [9]. Anti-HER2 therapies, such as trastuzumab and Pertuzumab, are primarily used as first-line treatment strategy, often in combination with adjunctive modalities including chemotherapies (doxorubicin-cyclophosphamide or docetaxel-carboplatin) [10,11] or CDK4/6 inhibitors that block cell cycle progression [12]. To extend the survival of metastatic HER2 BC patients, dual HER2 blockade with trastuzumab and Pertuzumab in combination with chemotherapy, primarily taxanes, is recommended [7,13]. Apart from that, trastuzumab emtansine (T-DM1) is typically utilized as the second-line therapy [14].
Additionally, triple-negative breast cancer (TNBC) is another more aggresstive subtype defined by the absence of ER, PR and HER2 expression. Based on gene profiling and cellular characteristics, TNBC can be further categorized into six molecular subtypes: basal-like 1 (BL1), basal-like 2 (BL2), mesenchymal, mesenchymal stem-like (MSL), immunomodulatory (IM), and luminal androgen receptor (LAR) [15]. In women with early TNBC, anthracycline- and taxane-based chemotherapy regimens serve as the cornerstone of treatment [14]. Moreover, the heterogeneity of TNBC contributes to its variable sensitivity to different therapeutics. For example, various mTOR or tyrosine kinase inhibitors can be used for M type TNBC [15,16] while anti-androgen receptor therapy is recommended for patients with LAR-subtype breast cancer [17]. Notably, Poly (ADP-ribose) polymerase inhibitors (PARPi) effectively disrupt DNA repair mechanisms and compromise genomic stability in BRCA1/2 mutant BC [18-20]. Additionally, immunotherapeutic approaches have gained attention, such as immune checkpoint inhibitor targeting PD-L1 on tumor cells or PD-1 on T cells. This strategy aims to prevent immune evasion and enhance T cells’ ability to recognize and destroy cancer cells [20-23].
Emerging evidence suggests that BC is composed not only of cancer cells but also an intricate tumor microenvironment (TME) that includes epithelial, stromal, and immune cells population. Within this TME, immune cells engage in complex interactions with cancer cells, mediated through both direct cellular contact and the release of soluble factors. These interactions collectively shape the microenvironment, thereby directly or indirectly influencing the responsiveness to anti-cancer therapies. For example, modulation of the anti-PD1-PDL1 axis has been demonstrated to directly enhance the tumoricidal activity of intratumoral T cells in BC, thereby increasing therapeutic efficacy [24]. Additionally, indirect mechanisms are prevalent; the infiltration of intratumoral T cells has been shown to significantly influence the response to BC chemotherapy [25], while the activity of immune checkpoint inhibitors can affect the success of anti-HER2 therapies in BC [26]. Furthermore, immune evasion and immunosuppression by tumor cells are evident through the secretion of bioactive factors (such as GM-CSF, IL-1β, VEGF, and PGE2), which act on immunoregulatory cells in the plasma, including myeloid-derived suppressor cells (MDSCs) and plasmacytoid dendritic cells (pDCs). These secreted factors can attenuate T cell cytotoxicity or indirectly reduce T cell infiltration, ultimately fostering cancer cell persistence [27].
The role of the immune microenvironment and associated immunological mechanisms in tumor modulation is inherently dual-faceted. The continuous and dynamic interplay between cancer cells and the immune microenvironment significantly influences cancer progression. Specifically, tumor-infiltrating immune cells can suppress tumor growth by targeting and eliminating immunogenic tumor cells. Conversely, these immune cells may also sculpt tumor immunogenicity, alter tumor differentiation cycles and characteristics, and potentially favor the selection of immune-tolerant cancer cell clones that can evade immune surveillance [25,28,29].
In conclusion, the immune microenvironment represents a pivotal element within tumors, closely associated with both tumor progression and therapeutic responses in cancer treatment. This review provides a comprehensive summary to illuminate how BC treatments interact with the immune landscape and aim to uncover the mechanisms that underlie and potentially enhance therapeutic efficacy.
2. Endocrine Therapy Impacts on the Immune System
Numerous studies have investigated the direct anti-tumor effects of endocrine therapies and related small molecule inhibitors. Given that various hormones bind to receptors on immune cells and exert biological effects [30,31] – and that cytokines from immune cells also influence the endocrine system [32], there is growing interest and evidence demonstrating a complex interplay between endocrine therapies and the tumor immune microenvironment. In this review, we comprehensively examine the immunomodulatory impacts of different endocrine therapeutic strategies (Figure 1).
2.1. Selective Estrogen Receptor Modulators (SERMs) – Tamoxifen
SERMs are drugs that bind to estrogen receptors and have tissue-specific effects, which act as either agonists or antagonists depending on the target tissue [33]. They are primarily used in the treatment and prevention of ER-positive breast cancer, where they inhibit estrogen-driven tumor growth. Although research is ongoing, the clinical use of SERMs is largely limited to BCs, with only limited evidence supporting their role in other gynecological cancers [34,35]. Common SERM drugs include but are not limited to tamoxifen (TX) and Ethamoxytriphetol (MER-25).
TX works by competitively binding to ER on breast cancer cells. Thus, it prevents estrogen from activating this signaling pathway that is needed to drive gene expression for cancer cell growth and division [36]. Beyond its direct anti-tumor effects, TX also has emerging controversial implications for the immune system. On one hand, evidence from animal models and in vitro experiments suggests that TX’s immune effects may occur through estrogen-independent pathways, such as P-glycoprotein inhibition, which sequentially affects dendritic cell function and favors Th2 responses while suppressing Th1 cytokine production [37, 38]. Additionally, TX-treated NK cells exhibit altered cytokine profiles and reduced the cytotoxicity, collectively reshaping the immune response toward a less cytotoxic profile [39]. On the other hand, TX was shown to enhance immune response by upregulating a series of interferon (IFN) and immune response genes, particularly within the JAK/STAT pathway in normal mammary epithelial cells [40]. Furthermore, TX boosts the cytotoxic activity of NK cells via upregulation of c-erbB-2 (HER2/neu) expression in HER2/neu nonamplified BCs, which leads to upregulation of ICAM-1 and increased tumor cell lysis through NK cell-mediated antibody-dependent cytotoxicity (ACDD) [41]. This dual role warrants further investigation on the immunomodulatory effects of TX in BCs.
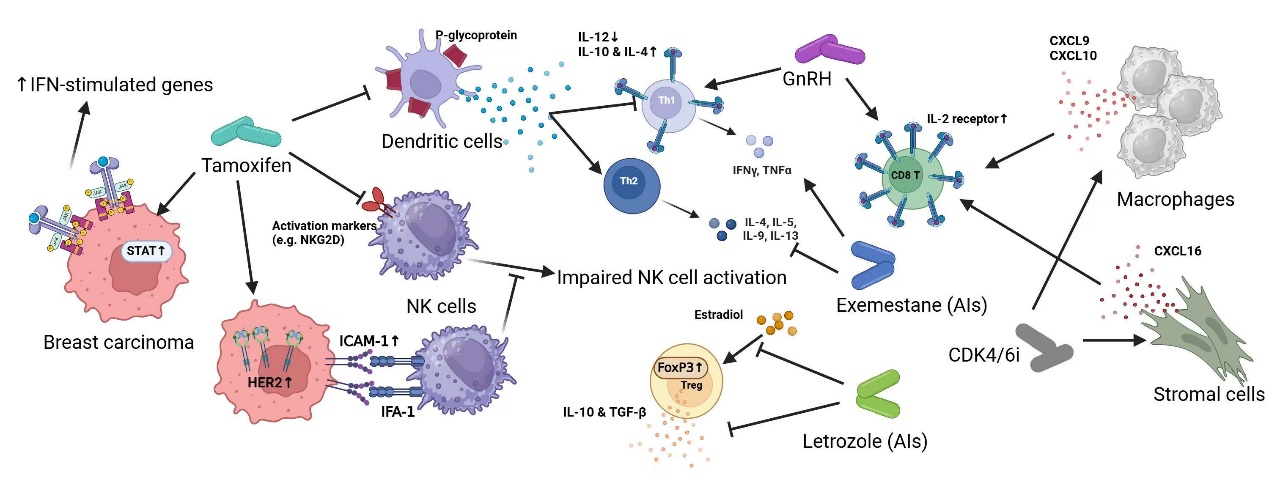
Figure 1: Endocrine therapy Impacts on the Immune system
2.2. Aromatase Inhibitors (AIs)
AIs are endocrine therapies that inhibit estrogen production, which blocks the enzyme aromatase, converting androgens into estrogens – a key estrogen source in postmenopausal women. Used mainly in postmenopausal patients with ER+ BC, AIs can be steroidal (e.g., exemestane) or non-steroidal (e.g., anastrozole, letrozole) [42,43]. Steroidal Ais binds irreversibly to the aromatase enzyme and permanently inactivate it, while non-steroidal Ais are reversible inhibitors that temporarily block the active site [43-49].
Evidence suggests that AIs may have complex effects on the immune system. Exemestane has been reported to affect immune cell populations, with some studies suggesting an increase in lymphocyte and a favorite shift in Th1 CD4+ T cells. This phenotypic observation has been implied to changes in bone metabolism and hormone levels [41,50]. Additionally, some evidence indicates that letrozole may reduce Treg cells and enhance immune surveillance against cancer [51,52]. However, AIs’ dual effect on immune function is notable: reducing estrogen levels may limit immune homeostasis, as estrogen is essential for T cell development and Th2 responses. Thus, while AIs may reduce immunosuppressive elements in the TME, they might also impair certain immune functions, such as extracellular pathogen defense, which highlights the need for further research on the immune impact AIs.
2.3. Ovarian Suppression (GnRH)
Ovarian suppression is frequently employed alongside chemotherapy and endocrine therapy, particularly effective in high-risk premenopausal patients. This therapy induces ovarian functional suppression to lower estrogen levels, achieved through luteinizing hormone-releasing hormone (LHRH) drugs or surgical ovarian removal [53,54]. Specifically, Gonadotropin-releasing hormone (GnRH) from hypothalamus, stimulates the pituitary gland to release gonadotropins, which regulate ovarian and testicular functions [55]. GnRH agonists are clinically used to suppress gonadotropins and reduce estrogen production, thus inhibiting the growth of hormone-dependent tumors.
Notably, GnRH and its agonists appear to influence the immune system in multiple ways. Research suggests that GnRH (I, II) may stimulate cytokines production (e.g., IFNγ, TNFβ, IL-3, IL-4, IL-7) in certain contexts [56,]. GnRH also directly influences immune cell function, such as by enhancing the expression of the IL-2 receptor (IL-2Rγ) on T-lymphocytes and B-lymphoblastoid cells, which can stimulate T cells and B cells proliferation and strengthen the immune system [57].
Interestingly, GnRH agonists exhibit a dual effect on hematopoietic stem cells: in Balb/c mice, prolonged GnRH agonist treatment reduces stem cell population and maturation rate in the bone marrow, whereas it increases the number and maturation activity in C57BL/6 mice [56, 57]. This strain-dependent variation highlights a complex, context-specific role of GnRH in immune modulation that warrants further study, especially regarding its clinical implications in humans.
2.4. Cyclin-Dependent Kinases 4 and 6 (CDK4/6) Inhibitors
CDK4/6i are commonly used alongside endocrine therapies in BC treatment, especially in ER+/HER2- BCs. CDK4/6 are cell cyclin-dependent kinases that bind with Cyclin D (D1, D2, D3 encoded by CCDN1, CCND2, and CCND3) to form CDK4/6-Cyclin D complexes. This complex phosphorylates the retinoblastoma protein (Rb), facilitating the transition from G1 phase (growth phase) to S phase (DNA synthesis phase) in the cell cycle [58]. CDK4/6 inhibitors prevent the formation of CDK4/6-Cyclin D complexes, thereby blocking Rb phosphorylation and arresting cancer cells in the G1 phase, which effectively slows tumor growth. In breast cancer therapy, CDK4/6 inhibitors (e.g., palbociclib, ribociclib, and abemaciclib) have shown clinical efficacy, especially in treating ER+/HER2- BC, making them a significant component of current therapeutic regimens.
It is also worth noting that CDK inhibitors impact the immune system. While CDK4/6 inhibitors may reduce T cell counts by blocking the G1-to-S phase transition in immune cells, cancer cells are shown to be more sensitive to CDK4/6 inhibition than T cells. Some CDK4/6i, such as Trilaciclib, may upregulate IL2 receptor components (IL2Rα, IL2Rβ, IL2Rγ), which potentially increases T cell counts and supports immune activation. Additionally, CDK4/6 inhibitors like abemaciclib can stimulate IL-2 secretion, and others, such as palbociclib and Trilaciclib, can enhance cytokines production (e.g., CXCL9, CXCL10, IFNγ, IL16, and CXCL16) to promote CTL and TH1 responses [59-61]. Furthermore, CDK4/6i may upregulate PD-L1 on tumor cells, making them more responsive to PD-1/PD-L1 antibodies (e.g., pembrolizumab, atezolizumab), which enhance T cell cytotoxicity [61,62]. This combination of CDK4/6 inhibitors with immune checkpoint inhibitors represents a promising therapeutic strategy for BC, especially TNBC, which warrants further investigation in clinical trials.
3. Anti-HER2 Therapy Impacts on the Immune System
Anti-HER2 therapy is a targeted therapeutic strategy that targets HER2 (human epidermal growth factor receptor 2), a transmembrane receptor frequently overexpressed in breast cancer, contributing to tumor progression and poor prognosis. By effectively binding on HER2 and suppressing HER2-driven signaling pathways, these therapies exert potent antitumor effects and have been widely used in both early-stage metastatic HER2+ BC patients [63]. Key anti-HER2 modalities include monoclonal antibodies, small-molecule tyrosine kinase inhibitors (TKIs), HER2 targeting antibody-drug conjugates (ADCs) and bispecific antibodies [64].
A growing body of preclinical and clinical evidence shows that anti-HER2 therapies, such as trastuzumab and other monoclonal antibodies, exerts significant effects on the immune system. Furthermore, the findings suggest that immune-related markers offer valuable predictive insights, and that immune system modulation may change clinical activity. This section will delve into the interplay between anti-HER2 therapy and the immune system, with a focus on the immunological mechanisms and their implications for therapeutic efficacy and clinical outcomes (Figure 2).
3.1. Anti-HER2 Antibody Therapy
Common anti-HER2 antibody drugs, such as Trastuzumab and Pertuzumab, inhibit HER2 signaling and enhance immune clearance. On one hand, Trastuzumab binds to domain IV of HER2, which blocks receptor dimerization and activating antibody-dependent cellular cytotoxicity (ADCC). On the other hand, Pertuzumab targets domain II of HER2 and prevents HER2-HER3 dimerization [65-67].
Trastuzumab exerts its therapeutic effects primarily through the innate immune system. By binding to HER2 on tumor cells and engaging FcγRIII (CD16) on NK cells, Trastuzumab activates Fc-FcR signaling to initiate ADCC. This mechanism enhances NK cells-mediated tumor cell lysis, ultimately promoting tumor regression [68,69]. While Trastuzumab’s role in directly modulating the adaptive immune response remains less prominent, Trastuzumab has been demonstrated to activate IL-12-IFN-γ axis, which bridges innate and adaptive immunity. By promoting the recruitment and activation of APCs and NK cells, Trastuzumab enhances IL-12 secretion by APCs, which in turn stimulates NK cells through IL-2 signaling to produce IFN-γ. IFN-γ further activates CD4+ T cells and facilitates their role in antitumor immunity. This cascade amplifies cellular immune responses and at the same time, damage-associated molecular patterns (DAMPs) releasing is also enhanced, which further activate immune pathways, such as FcR signaling [69-71].
Experimental evidence indicates that Trastuzumab increases a well-characterized DAMP HMGB1(High-mobility group box 1) levels in body fluids, which subsequently engages Toll-like receptors (TLRs) to trigger the MyD88-dependent signaling pathway [69,72].
In Trastuzumab-treated mouse models, a significant infiltration of CD8+ T cells was observed in the tumors, which underscores that antibody-induced immune activation within mediating antitumor effects plays a critical role. [69,71]. Additionally, anti-HER2 peptide vaccination strategies are notably which aim to combine Trastuzumab and HER2 peptides, such as E75 and GP2, to establish memory robust immune memory in patients. This combination therapy approach holds promise for enhancing long-term antitumor immunity and advancing vaccine-based treatments for HER2+ BCs [73,74].
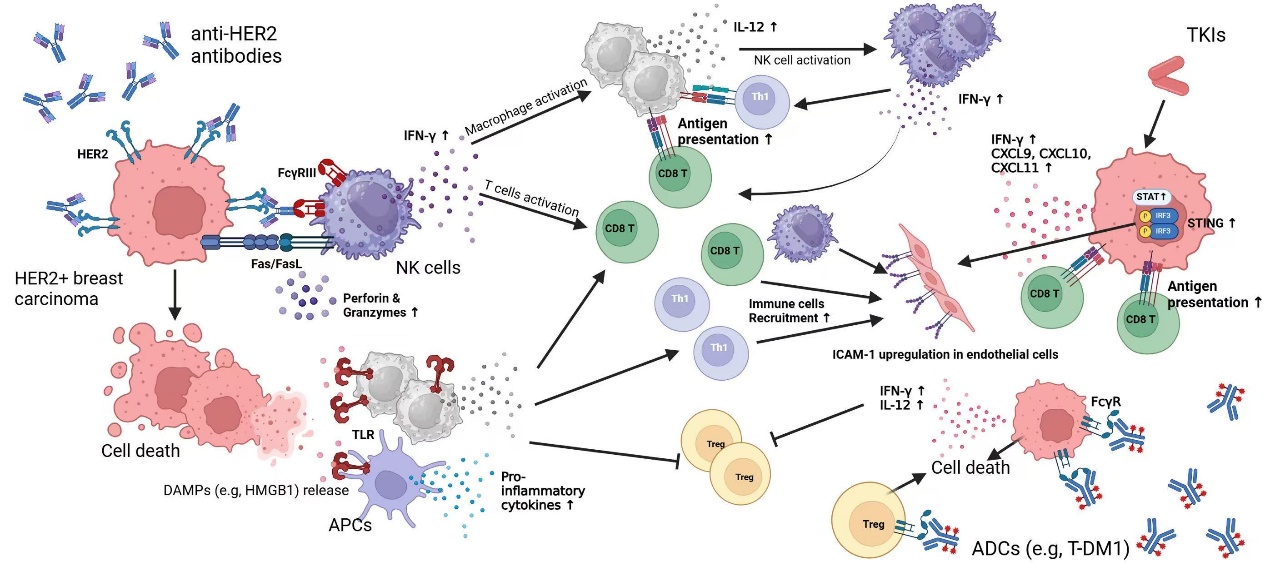
Figure 2: Anti-HER2 Antibody Therapy Impacts on the Immune System
3.2. Tyrosine Kinase Inhibitors (TKIs)
Common TKIs, including Lapatinib and Neratinib, function by competitively binding to the ATP-binding site of tyrosine kinases on HER2. This inhibits HER2 autophosphorylation and downstream signal transduction pathways, including the MAPK and PIK3 pathways (Lapatinib) [75], and ERK and Akt pathways (Neratinib) [6], thereby suppressing cancer cell growth [76-78].
Lapatinib not only disrupts erbB1/2 signaling but also modulates immune-related signaling pathways, which enhances antitumor immunity. Mechanistically, Lapatinib enhances CD8+ T cell activity by activating the Stat1 signaling pathway, which promotes CD8+ T cell infiltration into tumors and increases IFN-γ secretion. The central role of Stat1 is further highlighted by its regulation of chemokines such as CXCL9, CXCL10, and CXCL11, which facilitate T cell recruitment. Stat1 deficiency markedly reduced CD8+ T cell activation and tumor infiltration, which significantly impairs the antitumor efficacy of Lapatinib [79].
Stimulator of Interferon Genes (STING) is a key regulator of innate immunity, with the cGAS-STING signaling pathway in peripheral immune cells playing a central role in mediating inflammation and immune response. Through the detection of several inflammatory factors downstream the cGAS-STING cascade (IRF3, NF-κB p65), it was found that lapatinib can upregulate STING expression in GC tissue cells (SNU-216, MKN7 cells) by blocking the HER2 signaling pathway, while an increased number of CD8+ T cells were also detected [80,81].
Additionally, combining Lapatinib with Th1-derived cytokines has been shown to enhance cytotoxicity against HER2+ BCs through both synergistic and independent mechanisms. Lapatinib indirectly inhibits PI3K and AKT signaling to induce cancer cell apoptosis. Also, it has been demonstrated that Lapatinib can impact on the adaptive immune system by improving the sensitivity of immune cells to tumor cells. Lapatinib recruits a significant number of CD4+ and CD8+ cells, which secrete a large amount of Th1 cytokine IFN-γ. This, in turn, activates the STAT1 signaling pathway within the body, affecting the tumor microenvironment (TME) [75,82-84].
3.3. Antibody-Drug Conjugates (ADCs)
ADCs such as Kadcyla (T-DM1) and T-DXd combine HER2-targeting antibodies (e.g., Trastuzumab) with cytotoxic chemotherapeutic payloads (e.g., DM1 for T-DM1 and DXd for T-DXd). These therapies leverage the high specificity of HER2 antibodies to deliver cytotoxic agents directly to HER2-overexpressing tumor cells, which minimize off-target effects and enhance therapeutic efficacy [85-87].
Trastuzumab itself exerts immune-modulatory effects by engaging FcγRs and induce Fc-mediated ADCC. T-DM1 retains this capability through its Trastuzumab component, while its payload DM1 contributes additional antitumor effects by disrupting microtubules and modulating the TME [69,87].
In contrast to Trastuzumab, T-DM1 exhibits a distinctive ability to modulate the immune microenvironment system. On one hand, the toxicity of T-DM1 itself can decrease CD41+ and CD61+ cells. On the other hand, T-DM1 also readily binds to Fc, which further affects immune signaling pathways [87,88]. Notably, T-DM1 demonstrates enhanced efficacy in Trastuzumab-resistant models, which could partially be explained by its ability to reduce Tregs and suppress immune evasion by PD-L1 downregulation [89,90].
4. Other Treatments Impacting the Immune System
Ongoing research is shifting its focus from the development of novel therapeutics to the personalized treatment strategies tailored to the molecular characteristics of individual tumors. Several recently approved target therapies, including PARP inhibitors, PI3K inhibitors and mTOR inhibitors, demonstrate efficacy in defined biomarkers or genetic alterations. For instance, somatic PI3KCA mutations serve as predictive biomarkers for the response to PI3K inhibitors in ER+, HER2- metastatic BCs [91]. Similarly, germline BRCA mutations in metastatic BCs identify patients who potentially benefit from PARP inhibitor therapy [92]. Additionally, mTOR inhibitors have been shown to significantly extend progression-free survival, more than doubling its duration, in patients with ER+, HER2− endocrine-resistant metastatic breast cancer [93]. Given the expanding role of these targeted agents in breast cancer treatment, it is imperative to further investigate their immunomodulatory effects within the tumor microenvironment (TME) and explore their potential synergistic effects when combined with immunotherapies.
4.1. PI3K Inhibitor (Alpelisib, CYH33)
Alpelisib a PI3K inhibitor, is primarily utilized in treating HER2-, PIK3CA-mutant advanced breast cancer, often in combination with TX or other chemotherapeutic drugs. It inhibits PI3K (phosphoinositide 3-kinase) pathway, specifically targeting p110α, to suppress tumor cell proliferation and survival [94-95]. Alpelisib has been hypothesized to reduce T cells and DCs [96] and it has been associated with multiple immune-related adverse effects, such as cutaneous toxicity (rashes), GI disturbances (diarrhea), and hyperglycemia [97], while the underlying mechanism remains under investigation.
Interestingly, several mouse experiments found that Alpelisib can trigger a significant increase in CD8+ T cells [98]. This effect is attributed to the inhibition of PI3K pathway, which downregulates phosphatases (PTEN and PHLPP) and reduces the number and suppresses the function of Treg cells [99]. A similar immunomodulatory effect has been reported for CYH33 in mouse breast cancer models, where it has been shown to also promote fatty acid uptake and the recruitment of additional CD3+ T cells [100,101].
4.2. PARP Inhibitor (Olaparib)
Olaparib is a PARP inhibitor commonly used for the treatment of advanced breast cancer, particularly in BRCA mutation-associated TNBC or in patients with a high risk of recurrence following chemotherapy. It works by inhibiting the PARP enzyme, thereby disrupting DNA damage repair pathways in cancer cells and leading to genomic instability and tumor cell death [102].
Olaparib has a multifaceted impact on the immune system. In the BR5 tumor cell model, Olaparib treatment significantly increased CD8+ T cells and GzmB+ NK cells [103,104]. On the other hand, Olaparib can directly regulate immune-related gene expression. It has been shown to upregulate key genes involved in T cell infiltration and activation, including Cd8a, Zap70, Tbx21, Lck, Hck, Cd4, Fyn, Cd69, as well as genes related to cytotoxic T cell activity, such as Gzma and Prf1 [105]. Furthermore, Olaparib can activate certain immune pathways, particularly those involved in innate immune sensing and inflammatory responses. Notably, it enhances the type I IFN response, cGAS-STING pathway, and JAK/STAT pathway activation, these pathway regulations are suggested as secondary effects resulting from DNA damage accumulation [103,104,106].
Olaparib is also commonly used in combination with immune checkpoint inhibitors to enhance antitumor immunity. While Olaparib monotherapy may increase PD-L1 expression in cancer, combining with anti-PD-L1 therapy prevents immune evasion and strengthens CD8+ cell infiltration [104,105,107].
4.3. mTOR Inhibitor (Everolimus)
The mTOR signaling pathway is essential for T cell metabolism, proliferation and differentiation into different subsets, such as Th1, Th2, and Th17 cells [108]. In addition to regulating T cell fate, mTOR pathway controls immune metabolism, cytokine signaling, and antigen presentation. Notably, mTOR inhibition can impair cytokine (such as IL-4 and IL15) production, reduce NK cell activity and diminish recruitment of key immune proteins, such as recombination activating genes (RAGs) mediated by mTORC1 [109].
Everolimus (EVR) is an important mTOR-targeted inhibitor that suppresses cancer cell proliferation and angiogenesis [110,111]. While EVR shares immunosuppressive properties with other mTOR inhibitors, it does not directly reduce T cell apoptosis but instead inhibits their development and maturation, particularly in Karpas 299 and MyLa cell lines [112].
5. Future perspectives
The treatment landscape for breast cancer has evolved significantly. While these therapies primarily focus on tumor cell-intrinsic mechanisms, accumulating evidence highlights their immunomodulatory effects, which profoundly impact the TME and therapeutic response. Endocrine therapies, including SERMs, AIs, and ovarian suppression, modulate immune function by altering cytokine profiles, T cell activity, and antigen presentation. Similarly, CDK4/6 inhibitors influence the immune landscape by promoting T cell infiltration, upregulating immune-related genes, and modulating immune checkpoint expression.
In HER2-positive BCs, anti-HER2 monoclonal antibodies such as trastuzumab and pertuzumab not only inhibit oncogenic signaling but also enhance ADCC, which facilitates and connects innate and adaptive immune responses. Tyrosine kinase inhibitors like lapatinib further shape immune interactions by stimulating IFN-γ signaling and enhancing CD8+ T cell infiltration. Moreover, T-DM1 and novel immunotherapeutic combinations have demonstrated promising immune-enhancing effects, including PD-L1 downregulation and T cell activation.
Beyond conventional therapies, targeted agents such as PI3K inhibitors, PARP inhibitors, and mTOR inhibitors exhibit intricate immunomodulatory functions as well. These agents influence immune metabolism, T cell differentiation, and cytokine secretion, thereby altering tumor-immune interactions and potentially improving responses to immunotherapy.
Given the dynamic nature of the breast cancer immune microenvironment, there is a growing interest in combinatorial strategies that integrate targeted therapies with immune checkpoint inhibitors to enhance antitumor immunity. Future research should focus on elucidating the mechanistic basis of these immunomodulatory effects and optimizing therapeutic combinations to overcome immune resistance. A deeper understanding of how breast cancer therapies interact with the immune system will enable the development of more effective and personalized treatment approaches, ultimately improving clinical outcomes for breast cancer patients.
References
[1]. Siegel R L, Miller K D, Jemal A. Cancer statistics, 2019[J]. CA: a cancer journal for clinicians, 2019, 69(1): 7-34.
[2]. Hankinson S E, Colditz G A, Willett W C. Towards an integrated model for breast cancer etiology: the lifelong interplay of genes, lifestyle, and hormones[J]. Breast Cancer Research, 2004, 6: 1-6.
[3]. Danaei G, Vander Hoorn S, Lopez A D, et al. Causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors[J]. The lancet, 2005, 366(9499): 1784-1793.
[4]. Perou C M, Sørlie T, Eisen M B, et al. Molecular portraits of human breast tumours[J]. nature, 2000, 406(6797): 747-752.
[5]. Breast cancer Loibl, Sibylle et al. The Lancet, Volume 397, Issue 10286, 1750 - 1769
[6]. Prat A, Perou C M. Mammary development meets cancer genomics[J]. Nature medicine, 2009, 15(8): 842-844.
[7]. Burstein H J, Temin S, Anderson H, et al. Adjuvant endocrine therapy for women with hormone receptor–positive breast cancer: American Society of Clinical Oncology clinical practice guideline focused update[J]. Journal of clinical oncology, 2014, 32(21): 2255-2269.
[8]. Hicks D G, Kulkarni S. HER2+ breast cancer: review of biologic relevance and optimal use of diagnostic tools[J]. American journal of clinical pathology, 2008, 129(2): 263-273.
[9]. Yarden Y. Biology of HER2 and its importance in breast cancer[J]. Oncology, 2001, 61(Suppl. 2): 1-13.
[10]. Gianni L, Pienkowski T, Im Y H, et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): a multicentre, open-label, phase 2 randomised trial[J]. The lancet oncology, 2016, 17(6): 791-800.
[11]. Von Minckwitz G, Procter M, De Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer[J]. New England Journal of Medicine, 2017, 377(2): 122-131.
[12]. Corona S P, Ravelli A, Cretella D, et al. CDK4/6 inhibitors in HER2-positive breast cancer[J]. Critical reviews in oncology/hematology, 2017, 112: 208-214.
[13]. Xie B, Zhu L, Ma C, et al. A network meta-analysis on the efficacy of HER2-targeted agents in combination with taxane-containing regimens for treatment of HER2-positive metastatic breast cancer[J]. Breast Cancer, 2020, 27: 186-196.
[14]. Lehmann B D, Bauer J A, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies[J]. The Journal of clinical investigation, 2011, 121(7): 2750-2767.
[15]. Wang, DY., Jiang, Z., Ben-David, Y. et al. Molecular stratification within triple-negative breast cancer subtypes. Sci Rep 9, 19107 (2019).
[16]. He J, McLaughlin R P, van der Noord V, et al. Multi-targeted kinase inhibition alleviates mTOR inhibitor resistance in triple-negative breast cancer[J]. Breast Cancer Research and Treatment, 2019, 178: 263-274.
[17]. Vtorushin S, Dulesova A, Krakhmal N. Luminal androgen receptor (LAR) subtype of triple-negative breast cancer: molecular, morphological, and clinical features[J]. Journal of Zhejiang University-SCIENCE B, 2022, 23(8): 617-624.
[18]. Lee J M, Ledermann J A, Kohn E C. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies[J]. Annals of oncology, 2014, 25(1): 32-40.
[19]. Timms K M, Abkevich V, Hughes E, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes[J]. Breast Cancer Research, 2014, 16: 1-9.
[20]. Wen W X, Leong C O. Association of BRCA1-and BRCA2-deficiency with mutation burden, expression of PD-L1/PD-1, immune infiltrates, and T cell-inflamed signature in breast cancer[J]. PLoS One, 2019, 14(4): e0215381.
[21]. Kythreotou A, Siddique A, Mauri F A, et al. PD-L1[J]. Journal of clinical pathology, 2018, 71(3): 189-194.
[22]. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer[J]. American journal of cancer research, 2020, 10(3): 727.
[23]. Li C J, Lin L T, Hou M F, et al. PD‑L1/PD‑1 blockade in breast cancer: The immunotherapy era[J]. Oncology Reports, 2021, 45(1): 5-12.
[24]. Schütz F, Stefanovic S, Mayer L, et al. PD-1/PD-L1 pathway in breast cancer[J]. Oncology research and treatment, 2017, 40(5): 294-297.
[25]. Schmieder A, Michel J, Schönhaar K, et al. Differentiation and gene expression profile of tumor-associated macrophages[C]//Seminars in cancer biology. Academic Press, 2012, 22(4): 289-297.
[26]. Matusz-Fisher A, Tan A R. Combination of HER2-targeted agents with immune checkpoint inhibitors in the treatment of HER2-positive breast cancer[J]. Expert Opinion on Biological Therapy, 2022, 22(3): 385-395.
[27]. Vesely M D, Kershaw M H, Schreiber R D, et al. Natural innate and adaptive immunity to cancer[J]. Annual review of immunology, 2011, 29(1): 235-271.
[28]. Place A E, Jin Huh S, Polyak K. The microenvironment in breast cancer progression: biology and implications for treatment[J]. Breast cancer research, 2011, 13: 1-11.
[29]. Vesely M D, Kershaw M H, Schreiber R D, et al. Natural innate and adaptive immunity to cancer[J]. Annual review of immunology, 2011, 29(1): 235-271.
[30]. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways[J]. Cellular immunology, 2015, 294(2): 63-69.
[31]. Chakraborty B, Byemerwa J, Krebs T, et al. Estrogen receptor signaling in the immune system[J]. Endocrine reviews, 2023, 44(1): 117-141.
[32]. Silva C M, Isgaard J, Thorner M O. Cytokines in endocrine function[J]. Advances in protein chemistry, 1998, 52: 199-221.
[33]. Riggs B L, Hartmann L C. Selective estrogen-receptor modulators—mechanisms of action and application to clinical practice[J]. New England Journal of Medicine, 2003, 348(7): 618-629.
[34]. Chan K K L, Wei N, Liu S S, et al. Estrogen receptor subtypes in ovarian cancer: a clinical correlation[J]. Obstetrics & Gynecology, 2008, 111(1): 144-151.
[35]. Smith D, Stewart C J R, Clarke E M, et al. ER and PR expression and survival after endometrial cancer[J]. Gynecologic oncology, 2018, 148(2): 258-266.
[36]. Chang M. Tamoxifen resistance in breast cancer[J]. Biomolecules & therapeutics, 2012, 20(3): 256.
[37]. Behjati S, Frank M H. The effects of tamoxifen on immunity[J]. Current medicinal chemistry, 2009, 16(24): 3076-3080.
[38]. Bebo Jr B F, Dehghani B, Foster S, et al. Treatment with selective estrogen receptor modulators regulates myelin specific T‐cells and suppresses experimental autoimmune encephalomyelitis[J]. Glia, 2009, 57(7): 777-790.
[39]. Robinson E, Rubin D, Mekori T, et al. In vivo modulation of natural killer cell activity by tamoxifen in patients with bilateral primary breast cancer[J]. Cancer Immunology, Immunotherapy, 1993, 37: 209-212.
[40]. Schild-Hay L J, Leil T A, Divi R L, et al. Tamoxifen Induces Expression of Immune Response–Related Genes in Cultured Normal Human Mammary Epithelial Cells[J]. Cancer research, 2009, 69(3): 1150-1155.
[41]. Huang H, Zhou J, Chen H, et al. The immunomodulatory effects of endocrine therapy in breast cancer[J]. Journal of Experimental & Clinical Cancer Research, 2021, 40: 1-16.
[42]. Smith I E, Dowsett M. Aromatase inhibitors in breast cancer[J]. New England Journal of Medicine, 2003, 348(24): 2431-2442.
[43]. Chumsri S, Howes T, Bao T, et al. Aromatase, aromatase inhibitors, and breast cancer[J]. The Journal of steroid biochemistry and molecular biology, 2011, 125(1-2): 13-22.
[44]. Scott L J, Wiseman L R. Exemestane[J]. Drugs, 1999, 58: 675-680.
[45]. Pardhi M, Mehsram P, Bamgude P, et al. Synthesis of Chalcone Derivatives and its Biological Evaluations: An Overview[J]. 2025.
[46]. Kelly C M, Buzdar A U. Anastrozole[J]. Expert opinion on drug safety, 2010, 9(6): 995-1003.
[47]. Bhatnagar A S. The discovery and mechanism of action of letrozole[J]. Breast cancer research and treatment, 2007, 105(Suppl 1): 7-17.
[48]. Sabale P M, Sabale V P, Potey L C. Aromatase and aromatase inhibitors in breast cancer treatment[J]. J. Curr. Pharma Res., 2018, 9: 2636-2655.
[49]. Edris A, Abdelrahman M, Osman W, et al. Design of novel letrozole analogues targeting aromatase for breast cancer: molecular docking, molecular dynamics, and theoretical studies on gold nanoparticles[J]. Metabolites, 2023, 13(5): 583.
[50]. Zhao R, Chen C, Zhen L. Effects of zoledronic acid and exemestane combination therapy on immune function, sex hormones, bone markers, and clinical efficacy in hormone receptor-positive breast cancer patients[J]. European Journal of Gynaecological Oncology, 2024, 45(2).
[51]. Ma C X, Reinert T, Chmielewska I, et al. Mechanisms of aromatase inhibitor resistance[J]. Nature Reviews Cancer, 2015, 15(5): 261-275.
[52]. Generali D, Bates G, Berruti A, et al. Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients[J]. Clinical Cancer Research, 2009, 15(3): 1046-1051.
[53]. Bui K T, Willson M L, Goel S, et al. Ovarian suppression for adjuvant treatment of hormone receptor‐positive early breast cancer[J]. Cochrane Database of Systematic Reviews, 1996, 2020(3).
[54]. Francis P A, Regan M M, Fleming G F, et al. Adjuvant ovarian suppression in premenopausal breast cancer[J]. New England Journal of Medicine, 2015, 372(5): 436-446.
[55]. Millar R P. GnRHs and GnRH receptors[J]. Animal reproduction science, 2005, 88(1-2): 5-28.
[56]. Montgomery R A, Dallman M J. Analysis of cytokine gene expression during fetal thymic ontogeny using the polymerase chain reaction[J]. Journal of immunology (Baltimore, Md.: 1950), 1991, 147(2): 554-560.
[57]. Zakharova L, Sharova V, Izvolskaia M. Mechanisms of reciprocal regulation of gonadotropin-releasing hormone (GnRH)-producing and immune systems: the role of GnRH, cytokines and their receptors in early ontogenesis in normal and pathological conditions[J]. International journal of molecular sciences, 2020, 22(1): 114. receptors in early ontogenesis in normal and pathological conditions[J]. International Journal of Molecular Sciences, 2020, 22(1): 114.
[58]. Tanriverdi F, Gonzalez-Martinez D, Silveira L F G, et al. Expression of gonadotropin-releasing hormone type-I (GnRH-I) and type-II (GnRH-II) in human peripheral blood mononuclear cells (PMBCs) and regulation of B-lymphoblastoid cell proliferation by GnRH-I and GnRH-II[J]. Experimental and clinical endocrinology & diabetes, 2004, 112(10): 587-594. [59] Rao L V, Cleveland R P, Kimmel R J, et al. Hematopoietic Stem Cell Antigen‐1 (Sca‐1) Expression in Different Lymphoid Tissues of Female Mice Treated With GnRH Agonist[J]. American Journal of Reproductive Immunology, 1995, 34(4): 257-266.
[59]. Sherr C J, Beach D, Shapiro G I. Targeting CDK4 and CDK6: from discovery to therapy[J]. Cancer discovery, 2016, 6(4): 353-367.
[60]. Deng J, Wang E S, Jenkins R W, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation[J]. Cancer discovery, 2018, 8(2): 216-233.
[61]. Veinotte L, Gebremeskel S, Johnston B. CXCL16-positive dendritic cells enhance invariant natural killer T cell-dependent IFNγ production and tumor control[J]. Oncoimmunology, 2016, 5(6): e1160979.
[62]. Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy[J]. Nature, 2015, 527(7577): 249-253.
[63]. Patsoukis N, Wang Q, Strauss L, et al. Revisiting the PD-1 pathway[J]. Science advances, 2020, 6(38): eabd2712.
[64]. Schütz F, Stefanovic S, Mayer L, et al. PD-1/PD-L1 pathway in breast cancer[J]. Oncology research and treatment, 2017, 40(5): 294-297.
[65]. Ross J S, Slodkowska E A, Symmans W F, et al. The HER-2 receptor and breast cancer: ten years of targeted anti–HER-2 therapy and personalized medicine[J]. The oncologist, 2009, 14(4): 320-368.
[66]. Lopez-Albaitero A, Xu H, Guo H, et al. Overcoming resistance to HER2-targeted therapy with a novel HER2/CD3 bispecific antibody[J]. Oncoimmunology, 2017, 6(3): e1267891.
[67]. Boekhout A H, Beijnen J H, Schellens J H M. Trastuzumab[J]. The oncologist, 2011, 16(6): 800-810.
[68]. Capelan M, Pugliano L, De Azambuja E, et al. Pertuzumab: new hope for patients with HER2-positive breast cancer[J]. Annals of oncology, 2013, 24(2): 273-282.
[69]. Park J W, Kirpotin D B, Hong K, et al. Tumor targeting using anti-her2 immunoliposomes[J]. Journal of controlled release, 2001, 74(1-3): 95-113.
[70]. Musolino A, Gradishar W J, Rugo H S, et al. Role of Fcγ receptors in HER2-targeted breast cancer therapy[J]. Journal for immunotherapy of cancer, 2022, 10(1): e003171.
[71]. Park S G, Jiang Z, Mortenson E D, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity[J]. Cancer cell, 2010, 18(2): 160-170.
[72]. Jaime-Ramirez A C, Mundy-Bosse B L, Kondadasula S V, et al. IL-12 enhances the antitumor actions of trastuzumab via NK cell IFN-γ production[J]. The Journal of Immunology, 2011, 186(6): 3401-3409.
[73]. Vignali D A A, Kuchroo V K. IL-12 family cytokines: immunological playmakers[J]. Nature immunology, 2012, 13(8): 722-728.
[74]. Deguine J, Barton G M. MyD88: a central player in innate immune signaling[J]. F1000prime reports, 2014, 6: 97.
[75]. Nuti M, Bellati F, Visconti V, et al. Immune effects of trastuzumab[J]. Journal of Cancer, 2011, 2: 317.
[76]. Benavides L C, Sears A K, Gates J D, et al. Comparison of different HER2/neu vaccines in adjuvant breast cancer trials: implications for dosing of peptide vaccines[J]. Expert review of vaccines, 2011, 10(2): 201-210.
[77]. Schlam I, Swain S M. HER2-positive breast cancer and tyrosine kinase inhibitors: the time is now[J]. NPJ breast cancer, 2021, 7(1): 56.
[78]. Xuhong J C, Qi X W, Zhang Y, et al. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer[J]. American journal of cancer research, 2019, 9(10): 2103.
[79]. Gotink K J, Verheul H M W. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action?[J]. Angiogenesis, 2010, 13: 1-14.
[80]. Garrett J T, Arteaga C L. Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: mechanisms and clinical implications[J]. Cancer biology & therapy, 2011, 11(9): 793-800.
[81]. Hannesdóttir L, Tymoszuk P, Parajuli N, et al. Lapatinib and doxorubicin enhance the S tat1‐dependent antitumor immune response[J]. European journal of immunology, 2013, 43(10): 2718-2729.
[82]. Fukai S, Nakajima S, Saito M, et al. Down-regulation of stimulator of interferon genes (STING) expression and CD8+ T-cell infiltration depending on HER2 heterogeneity in HER2-positive gastric cancer[J]. Gastric Cancer, 2023, 26(6): 878-890.
[83]. Hopfner K P, Hornung V. Molecular mechanisms and cellular functions of cGAS–STING signalling[J]. Nature reviews Molecular cell biology, 2020, 21(9): 501-521.
[84]. Showalter L E, Oechsle C, Ghimirey N, et al. Th1 cytokines sensitize HER-expressing breast cancer cells to lapatinib[J]. PLoS One, 2019, 14(1): e0210209.
[85]. Hannesdóttir L, Tymoszuk P, Parajuli N, et al. Lapatinib and doxorubicin enhance the S tat1‐dependent antitumor immune response[J]. European journal of immunology, 2013, 43(10): 2718-2729.
[86]. Rinnerthaler G, Gampenrieder S P, Greil R. HER2 directed antibody-drug-conjugates beyond T-DM1 in breast cancer[J]. International journal of molecular sciences, 2019, 20(5): 1115.
[87]. Krop I, Winer E P. Trastuzumab emtansine: a novel antibody–drug conjugate for HER2-positive breast cancer[J]. Clinical Cancer Research, 2014, 20(1): 15-20.
[88]. Hurvitz S, Kim S B, Chung W P, et al. Abstract GS3-01: Trastuzumab deruxtecan (T-DXd; DS-8201a) vs. trastuzumab emtansine (T-DM1) in patients (pts) with HER2+ metastatic breast cancer (mBC): subgroup analyses from the randomized phase 3 study DESTINY-Breast03[J]. Cancer Research, 2022, 82(4_Supplement): GS3-01-GS3-01.
[89]. Uppal H, Doudement E, Mahapatra K, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1)[J]. Clinical Cancer Research, 2015, 21(1): 123-133.
[90]. Liu F, Mao K, Chen H, et al. Enhancing the Safety and Efficacy of Trastuzumab Emtansine (T‐DM1) Through Nano‐Delivery System in Breast Cancer Therapy[J]. Small, 2024, 20(50): 2400977.
[91]. Hunter F W, Barker H R, Lipert B, et al. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer[J]. British journal of cancer, 2020, 122(5): 603-612.
[92]. Kim R. Cancer immunoediting: from immune surveillance to immune escape[J]. Cancer Immunotherapy, 2007: 9-27.
[93]. André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer[J]. New England Journal of Medicine, 2019, 380(20): 1929-1940.
[94]. Robson M, Im S A, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation[J]. New England Journal of Medicine, 2017, 377(6): 523-533.
[95]. Piccart M, Hortobagyi G N, Campone M, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2[J]. Annals of oncology, 2014, 25(12): 2357-2362.
[96]. Yan C, Yang J, Saleh N, et al. Inhibition of the PI3K/mTOR pathway in breast cancer to enhance response to immune checkpoint inhibitors in breast cancer[J]. International journal of molecular sciences, 2021, 22(10): 5207.
[97]. André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer[J]. New England Journal of Medicine, 2019, 380(20): 1929-1940.
[98]. Pompura S L, Dominguez-Villar M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function[J]. Journal of leukocyte biology, 2018, 103(6): 1065-1076.
[99]. Armaghani A J, Han H S. Alpelisib in the treatment of breast cancer: a short review on the emerging clinical data[J]. Breast Cancer: Targets and Therapy, 2020: 251-258.
[100]. Li X, Chen G, Wang F, et al. Oncogenic PIK3CA recruits myeloid‐derived suppressor cells to shape the immunosuppressive tumour microenvironment in luminal breast cancer through the 5‐lipoxygenase‐dependent arachidonic acid pathway[J]. Clinical and Translational Medicine, 2023, 13(11): e1483.
[101]. Stark A K, Davenport E, Patton D T, et al. Loss of phosphatidylinositol 3-kinase activity in regulatory T cells leads to neuronal inflammation[J]. The Journal of Immunology, 2020, 205(1): 78-89.
[102]. Sun P, Zhang X, Wang R J, et al. PI3Kα inhibitor CYH33 triggers antitumor immunity in murine breast cancer by activating CD8+ T cells and promoting fatty acid metabolism[J]. Journal for ImmunoTherapy of Cancer, 2021, 9(8): e003093.
[103]. Drullinsky P R, Hurvitz S A. Mechanistic basis for PI3K inhibitor antitumor activity and adverse reactions in advanced breast cancer[J]. Breast cancer research and treatment, 2020, 181: 233-248.
[104]. Griguolo G, Dieci M V, Guarneri V, et al. Olaparib for the treatment of breast cancer[J]. Expert review of anticancer therapy, 2018, 18(6): 519-530.
[105]. Staniszewska A D, Armenia J, King M, et al. PARP inhibition is a modulator of anti-tumor immune response in BRCA-deficient tumors[J]. Oncoimmunology, 2022, 11(1): 2083755.
[106]. Pantelidou C, Sonzogni O, De Oliveria Taveira M, et al. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer[J]. Cancer discovery, 2019, 9(6): 722-737.
[107]. Pantelidou C, Sonzogni O, De Oliveria Taveira M, et al. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer[J]. Cancer discovery, 2019, 9(6): 722-737.
[108]. Lampert E J, Zimmer A, Padget M, et al. Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: a proof-of-concept phase II study[J]. Clinical Cancer Research, 2020, 26(16): 4268-4279.
[109]. Jiao S, Xia W, Yamaguchi H, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression[J]. Clinical Cancer Research, 2017, 23(14): 3711-3720.
[110]. Powell J D, Pollizzi K N, Heikamp E B, et al. Regulation of immune responses by mTOR[J]. Annual review of immunology, 2012, 30(1): 39-68.
[111]. Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR[J]. Nature Reviews Immunology, 2015, 15(10): 599-614.
[112]. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer[J]. New England Journal of Medicine, 2012, 366(6): 520-529.
[113]. Hare S H, Harvey A J. mTOR function and therapeutic targeting in breast cancer[J]. American journal of cancer research, 2017, 7(3): 383.
[114]. Witzig T E, Reeder C, Han J J, et al. The mTORC1 inhibitor everolimus has antitumor activity in vitro and produces tumor responses in patients with relapsed T-cell lymphoma[J]. Blood, The Journal of the American Society of Hematology, 2015, 126(3): 328-335.
Cite this article
Xu,R. (2025). Immunomodulatory Effects of Breast Cancer Therapies: Unraveling the Crosstalk Between Therapeutics and the Tumor Immune Microenvironment. Theoretical and Natural Science,99,51-63.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 5th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Siegel R L, Miller K D, Jemal A. Cancer statistics, 2019[J]. CA: a cancer journal for clinicians, 2019, 69(1): 7-34.
[2]. Hankinson S E, Colditz G A, Willett W C. Towards an integrated model for breast cancer etiology: the lifelong interplay of genes, lifestyle, and hormones[J]. Breast Cancer Research, 2004, 6: 1-6.
[3]. Danaei G, Vander Hoorn S, Lopez A D, et al. Causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors[J]. The lancet, 2005, 366(9499): 1784-1793.
[4]. Perou C M, Sørlie T, Eisen M B, et al. Molecular portraits of human breast tumours[J]. nature, 2000, 406(6797): 747-752.
[5]. Breast cancer Loibl, Sibylle et al. The Lancet, Volume 397, Issue 10286, 1750 - 1769
[6]. Prat A, Perou C M. Mammary development meets cancer genomics[J]. Nature medicine, 2009, 15(8): 842-844.
[7]. Burstein H J, Temin S, Anderson H, et al. Adjuvant endocrine therapy for women with hormone receptor–positive breast cancer: American Society of Clinical Oncology clinical practice guideline focused update[J]. Journal of clinical oncology, 2014, 32(21): 2255-2269.
[8]. Hicks D G, Kulkarni S. HER2+ breast cancer: review of biologic relevance and optimal use of diagnostic tools[J]. American journal of clinical pathology, 2008, 129(2): 263-273.
[9]. Yarden Y. Biology of HER2 and its importance in breast cancer[J]. Oncology, 2001, 61(Suppl. 2): 1-13.
[10]. Gianni L, Pienkowski T, Im Y H, et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): a multicentre, open-label, phase 2 randomised trial[J]. The lancet oncology, 2016, 17(6): 791-800.
[11]. Von Minckwitz G, Procter M, De Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer[J]. New England Journal of Medicine, 2017, 377(2): 122-131.
[12]. Corona S P, Ravelli A, Cretella D, et al. CDK4/6 inhibitors in HER2-positive breast cancer[J]. Critical reviews in oncology/hematology, 2017, 112: 208-214.
[13]. Xie B, Zhu L, Ma C, et al. A network meta-analysis on the efficacy of HER2-targeted agents in combination with taxane-containing regimens for treatment of HER2-positive metastatic breast cancer[J]. Breast Cancer, 2020, 27: 186-196.
[14]. Lehmann B D, Bauer J A, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies[J]. The Journal of clinical investigation, 2011, 121(7): 2750-2767.
[15]. Wang, DY., Jiang, Z., Ben-David, Y. et al. Molecular stratification within triple-negative breast cancer subtypes. Sci Rep 9, 19107 (2019).
[16]. He J, McLaughlin R P, van der Noord V, et al. Multi-targeted kinase inhibition alleviates mTOR inhibitor resistance in triple-negative breast cancer[J]. Breast Cancer Research and Treatment, 2019, 178: 263-274.
[17]. Vtorushin S, Dulesova A, Krakhmal N. Luminal androgen receptor (LAR) subtype of triple-negative breast cancer: molecular, morphological, and clinical features[J]. Journal of Zhejiang University-SCIENCE B, 2022, 23(8): 617-624.
[18]. Lee J M, Ledermann J A, Kohn E C. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies[J]. Annals of oncology, 2014, 25(1): 32-40.
[19]. Timms K M, Abkevich V, Hughes E, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes[J]. Breast Cancer Research, 2014, 16: 1-9.
[20]. Wen W X, Leong C O. Association of BRCA1-and BRCA2-deficiency with mutation burden, expression of PD-L1/PD-1, immune infiltrates, and T cell-inflamed signature in breast cancer[J]. PLoS One, 2019, 14(4): e0215381.
[21]. Kythreotou A, Siddique A, Mauri F A, et al. PD-L1[J]. Journal of clinical pathology, 2018, 71(3): 189-194.
[22]. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer[J]. American journal of cancer research, 2020, 10(3): 727.
[23]. Li C J, Lin L T, Hou M F, et al. PD‑L1/PD‑1 blockade in breast cancer: The immunotherapy era[J]. Oncology Reports, 2021, 45(1): 5-12.
[24]. Schütz F, Stefanovic S, Mayer L, et al. PD-1/PD-L1 pathway in breast cancer[J]. Oncology research and treatment, 2017, 40(5): 294-297.
[25]. Schmieder A, Michel J, Schönhaar K, et al. Differentiation and gene expression profile of tumor-associated macrophages[C]//Seminars in cancer biology. Academic Press, 2012, 22(4): 289-297.
[26]. Matusz-Fisher A, Tan A R. Combination of HER2-targeted agents with immune checkpoint inhibitors in the treatment of HER2-positive breast cancer[J]. Expert Opinion on Biological Therapy, 2022, 22(3): 385-395.
[27]. Vesely M D, Kershaw M H, Schreiber R D, et al. Natural innate and adaptive immunity to cancer[J]. Annual review of immunology, 2011, 29(1): 235-271.
[28]. Place A E, Jin Huh S, Polyak K. The microenvironment in breast cancer progression: biology and implications for treatment[J]. Breast cancer research, 2011, 13: 1-11.
[29]. Vesely M D, Kershaw M H, Schreiber R D, et al. Natural innate and adaptive immunity to cancer[J]. Annual review of immunology, 2011, 29(1): 235-271.
[30]. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways[J]. Cellular immunology, 2015, 294(2): 63-69.
[31]. Chakraborty B, Byemerwa J, Krebs T, et al. Estrogen receptor signaling in the immune system[J]. Endocrine reviews, 2023, 44(1): 117-141.
[32]. Silva C M, Isgaard J, Thorner M O. Cytokines in endocrine function[J]. Advances in protein chemistry, 1998, 52: 199-221.
[33]. Riggs B L, Hartmann L C. Selective estrogen-receptor modulators—mechanisms of action and application to clinical practice[J]. New England Journal of Medicine, 2003, 348(7): 618-629.
[34]. Chan K K L, Wei N, Liu S S, et al. Estrogen receptor subtypes in ovarian cancer: a clinical correlation[J]. Obstetrics & Gynecology, 2008, 111(1): 144-151.
[35]. Smith D, Stewart C J R, Clarke E M, et al. ER and PR expression and survival after endometrial cancer[J]. Gynecologic oncology, 2018, 148(2): 258-266.
[36]. Chang M. Tamoxifen resistance in breast cancer[J]. Biomolecules & therapeutics, 2012, 20(3): 256.
[37]. Behjati S, Frank M H. The effects of tamoxifen on immunity[J]. Current medicinal chemistry, 2009, 16(24): 3076-3080.
[38]. Bebo Jr B F, Dehghani B, Foster S, et al. Treatment with selective estrogen receptor modulators regulates myelin specific T‐cells and suppresses experimental autoimmune encephalomyelitis[J]. Glia, 2009, 57(7): 777-790.
[39]. Robinson E, Rubin D, Mekori T, et al. In vivo modulation of natural killer cell activity by tamoxifen in patients with bilateral primary breast cancer[J]. Cancer Immunology, Immunotherapy, 1993, 37: 209-212.
[40]. Schild-Hay L J, Leil T A, Divi R L, et al. Tamoxifen Induces Expression of Immune Response–Related Genes in Cultured Normal Human Mammary Epithelial Cells[J]. Cancer research, 2009, 69(3): 1150-1155.
[41]. Huang H, Zhou J, Chen H, et al. The immunomodulatory effects of endocrine therapy in breast cancer[J]. Journal of Experimental & Clinical Cancer Research, 2021, 40: 1-16.
[42]. Smith I E, Dowsett M. Aromatase inhibitors in breast cancer[J]. New England Journal of Medicine, 2003, 348(24): 2431-2442.
[43]. Chumsri S, Howes T, Bao T, et al. Aromatase, aromatase inhibitors, and breast cancer[J]. The Journal of steroid biochemistry and molecular biology, 2011, 125(1-2): 13-22.
[44]. Scott L J, Wiseman L R. Exemestane[J]. Drugs, 1999, 58: 675-680.
[45]. Pardhi M, Mehsram P, Bamgude P, et al. Synthesis of Chalcone Derivatives and its Biological Evaluations: An Overview[J]. 2025.
[46]. Kelly C M, Buzdar A U. Anastrozole[J]. Expert opinion on drug safety, 2010, 9(6): 995-1003.
[47]. Bhatnagar A S. The discovery and mechanism of action of letrozole[J]. Breast cancer research and treatment, 2007, 105(Suppl 1): 7-17.
[48]. Sabale P M, Sabale V P, Potey L C. Aromatase and aromatase inhibitors in breast cancer treatment[J]. J. Curr. Pharma Res., 2018, 9: 2636-2655.
[49]. Edris A, Abdelrahman M, Osman W, et al. Design of novel letrozole analogues targeting aromatase for breast cancer: molecular docking, molecular dynamics, and theoretical studies on gold nanoparticles[J]. Metabolites, 2023, 13(5): 583.
[50]. Zhao R, Chen C, Zhen L. Effects of zoledronic acid and exemestane combination therapy on immune function, sex hormones, bone markers, and clinical efficacy in hormone receptor-positive breast cancer patients[J]. European Journal of Gynaecological Oncology, 2024, 45(2).
[51]. Ma C X, Reinert T, Chmielewska I, et al. Mechanisms of aromatase inhibitor resistance[J]. Nature Reviews Cancer, 2015, 15(5): 261-275.
[52]. Generali D, Bates G, Berruti A, et al. Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients[J]. Clinical Cancer Research, 2009, 15(3): 1046-1051.
[53]. Bui K T, Willson M L, Goel S, et al. Ovarian suppression for adjuvant treatment of hormone receptor‐positive early breast cancer[J]. Cochrane Database of Systematic Reviews, 1996, 2020(3).
[54]. Francis P A, Regan M M, Fleming G F, et al. Adjuvant ovarian suppression in premenopausal breast cancer[J]. New England Journal of Medicine, 2015, 372(5): 436-446.
[55]. Millar R P. GnRHs and GnRH receptors[J]. Animal reproduction science, 2005, 88(1-2): 5-28.
[56]. Montgomery R A, Dallman M J. Analysis of cytokine gene expression during fetal thymic ontogeny using the polymerase chain reaction[J]. Journal of immunology (Baltimore, Md.: 1950), 1991, 147(2): 554-560.
[57]. Zakharova L, Sharova V, Izvolskaia M. Mechanisms of reciprocal regulation of gonadotropin-releasing hormone (GnRH)-producing and immune systems: the role of GnRH, cytokines and their receptors in early ontogenesis in normal and pathological conditions[J]. International journal of molecular sciences, 2020, 22(1): 114. receptors in early ontogenesis in normal and pathological conditions[J]. International Journal of Molecular Sciences, 2020, 22(1): 114.
[58]. Tanriverdi F, Gonzalez-Martinez D, Silveira L F G, et al. Expression of gonadotropin-releasing hormone type-I (GnRH-I) and type-II (GnRH-II) in human peripheral blood mononuclear cells (PMBCs) and regulation of B-lymphoblastoid cell proliferation by GnRH-I and GnRH-II[J]. Experimental and clinical endocrinology & diabetes, 2004, 112(10): 587-594. [59] Rao L V, Cleveland R P, Kimmel R J, et al. Hematopoietic Stem Cell Antigen‐1 (Sca‐1) Expression in Different Lymphoid Tissues of Female Mice Treated With GnRH Agonist[J]. American Journal of Reproductive Immunology, 1995, 34(4): 257-266.
[59]. Sherr C J, Beach D, Shapiro G I. Targeting CDK4 and CDK6: from discovery to therapy[J]. Cancer discovery, 2016, 6(4): 353-367.
[60]. Deng J, Wang E S, Jenkins R W, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation[J]. Cancer discovery, 2018, 8(2): 216-233.
[61]. Veinotte L, Gebremeskel S, Johnston B. CXCL16-positive dendritic cells enhance invariant natural killer T cell-dependent IFNγ production and tumor control[J]. Oncoimmunology, 2016, 5(6): e1160979.
[62]. Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy[J]. Nature, 2015, 527(7577): 249-253.
[63]. Patsoukis N, Wang Q, Strauss L, et al. Revisiting the PD-1 pathway[J]. Science advances, 2020, 6(38): eabd2712.
[64]. Schütz F, Stefanovic S, Mayer L, et al. PD-1/PD-L1 pathway in breast cancer[J]. Oncology research and treatment, 2017, 40(5): 294-297.
[65]. Ross J S, Slodkowska E A, Symmans W F, et al. The HER-2 receptor and breast cancer: ten years of targeted anti–HER-2 therapy and personalized medicine[J]. The oncologist, 2009, 14(4): 320-368.
[66]. Lopez-Albaitero A, Xu H, Guo H, et al. Overcoming resistance to HER2-targeted therapy with a novel HER2/CD3 bispecific antibody[J]. Oncoimmunology, 2017, 6(3): e1267891.
[67]. Boekhout A H, Beijnen J H, Schellens J H M. Trastuzumab[J]. The oncologist, 2011, 16(6): 800-810.
[68]. Capelan M, Pugliano L, De Azambuja E, et al. Pertuzumab: new hope for patients with HER2-positive breast cancer[J]. Annals of oncology, 2013, 24(2): 273-282.
[69]. Park J W, Kirpotin D B, Hong K, et al. Tumor targeting using anti-her2 immunoliposomes[J]. Journal of controlled release, 2001, 74(1-3): 95-113.
[70]. Musolino A, Gradishar W J, Rugo H S, et al. Role of Fcγ receptors in HER2-targeted breast cancer therapy[J]. Journal for immunotherapy of cancer, 2022, 10(1): e003171.
[71]. Park S G, Jiang Z, Mortenson E D, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity[J]. Cancer cell, 2010, 18(2): 160-170.
[72]. Jaime-Ramirez A C, Mundy-Bosse B L, Kondadasula S V, et al. IL-12 enhances the antitumor actions of trastuzumab via NK cell IFN-γ production[J]. The Journal of Immunology, 2011, 186(6): 3401-3409.
[73]. Vignali D A A, Kuchroo V K. IL-12 family cytokines: immunological playmakers[J]. Nature immunology, 2012, 13(8): 722-728.
[74]. Deguine J, Barton G M. MyD88: a central player in innate immune signaling[J]. F1000prime reports, 2014, 6: 97.
[75]. Nuti M, Bellati F, Visconti V, et al. Immune effects of trastuzumab[J]. Journal of Cancer, 2011, 2: 317.
[76]. Benavides L C, Sears A K, Gates J D, et al. Comparison of different HER2/neu vaccines in adjuvant breast cancer trials: implications for dosing of peptide vaccines[J]. Expert review of vaccines, 2011, 10(2): 201-210.
[77]. Schlam I, Swain S M. HER2-positive breast cancer and tyrosine kinase inhibitors: the time is now[J]. NPJ breast cancer, 2021, 7(1): 56.
[78]. Xuhong J C, Qi X W, Zhang Y, et al. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer[J]. American journal of cancer research, 2019, 9(10): 2103.
[79]. Gotink K J, Verheul H M W. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action?[J]. Angiogenesis, 2010, 13: 1-14.
[80]. Garrett J T, Arteaga C L. Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: mechanisms and clinical implications[J]. Cancer biology & therapy, 2011, 11(9): 793-800.
[81]. Hannesdóttir L, Tymoszuk P, Parajuli N, et al. Lapatinib and doxorubicin enhance the S tat1‐dependent antitumor immune response[J]. European journal of immunology, 2013, 43(10): 2718-2729.
[82]. Fukai S, Nakajima S, Saito M, et al. Down-regulation of stimulator of interferon genes (STING) expression and CD8+ T-cell infiltration depending on HER2 heterogeneity in HER2-positive gastric cancer[J]. Gastric Cancer, 2023, 26(6): 878-890.
[83]. Hopfner K P, Hornung V. Molecular mechanisms and cellular functions of cGAS–STING signalling[J]. Nature reviews Molecular cell biology, 2020, 21(9): 501-521.
[84]. Showalter L E, Oechsle C, Ghimirey N, et al. Th1 cytokines sensitize HER-expressing breast cancer cells to lapatinib[J]. PLoS One, 2019, 14(1): e0210209.
[85]. Hannesdóttir L, Tymoszuk P, Parajuli N, et al. Lapatinib and doxorubicin enhance the S tat1‐dependent antitumor immune response[J]. European journal of immunology, 2013, 43(10): 2718-2729.
[86]. Rinnerthaler G, Gampenrieder S P, Greil R. HER2 directed antibody-drug-conjugates beyond T-DM1 in breast cancer[J]. International journal of molecular sciences, 2019, 20(5): 1115.
[87]. Krop I, Winer E P. Trastuzumab emtansine: a novel antibody–drug conjugate for HER2-positive breast cancer[J]. Clinical Cancer Research, 2014, 20(1): 15-20.
[88]. Hurvitz S, Kim S B, Chung W P, et al. Abstract GS3-01: Trastuzumab deruxtecan (T-DXd; DS-8201a) vs. trastuzumab emtansine (T-DM1) in patients (pts) with HER2+ metastatic breast cancer (mBC): subgroup analyses from the randomized phase 3 study DESTINY-Breast03[J]. Cancer Research, 2022, 82(4_Supplement): GS3-01-GS3-01.
[89]. Uppal H, Doudement E, Mahapatra K, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1)[J]. Clinical Cancer Research, 2015, 21(1): 123-133.
[90]. Liu F, Mao K, Chen H, et al. Enhancing the Safety and Efficacy of Trastuzumab Emtansine (T‐DM1) Through Nano‐Delivery System in Breast Cancer Therapy[J]. Small, 2024, 20(50): 2400977.
[91]. Hunter F W, Barker H R, Lipert B, et al. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer[J]. British journal of cancer, 2020, 122(5): 603-612.
[92]. Kim R. Cancer immunoediting: from immune surveillance to immune escape[J]. Cancer Immunotherapy, 2007: 9-27.
[93]. André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer[J]. New England Journal of Medicine, 2019, 380(20): 1929-1940.
[94]. Robson M, Im S A, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation[J]. New England Journal of Medicine, 2017, 377(6): 523-533.
[95]. Piccart M, Hortobagyi G N, Campone M, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2[J]. Annals of oncology, 2014, 25(12): 2357-2362.
[96]. Yan C, Yang J, Saleh N, et al. Inhibition of the PI3K/mTOR pathway in breast cancer to enhance response to immune checkpoint inhibitors in breast cancer[J]. International journal of molecular sciences, 2021, 22(10): 5207.
[97]. André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer[J]. New England Journal of Medicine, 2019, 380(20): 1929-1940.
[98]. Pompura S L, Dominguez-Villar M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function[J]. Journal of leukocyte biology, 2018, 103(6): 1065-1076.
[99]. Armaghani A J, Han H S. Alpelisib in the treatment of breast cancer: a short review on the emerging clinical data[J]. Breast Cancer: Targets and Therapy, 2020: 251-258.
[100]. Li X, Chen G, Wang F, et al. Oncogenic PIK3CA recruits myeloid‐derived suppressor cells to shape the immunosuppressive tumour microenvironment in luminal breast cancer through the 5‐lipoxygenase‐dependent arachidonic acid pathway[J]. Clinical and Translational Medicine, 2023, 13(11): e1483.
[101]. Stark A K, Davenport E, Patton D T, et al. Loss of phosphatidylinositol 3-kinase activity in regulatory T cells leads to neuronal inflammation[J]. The Journal of Immunology, 2020, 205(1): 78-89.
[102]. Sun P, Zhang X, Wang R J, et al. PI3Kα inhibitor CYH33 triggers antitumor immunity in murine breast cancer by activating CD8+ T cells and promoting fatty acid metabolism[J]. Journal for ImmunoTherapy of Cancer, 2021, 9(8): e003093.
[103]. Drullinsky P R, Hurvitz S A. Mechanistic basis for PI3K inhibitor antitumor activity and adverse reactions in advanced breast cancer[J]. Breast cancer research and treatment, 2020, 181: 233-248.
[104]. Griguolo G, Dieci M V, Guarneri V, et al. Olaparib for the treatment of breast cancer[J]. Expert review of anticancer therapy, 2018, 18(6): 519-530.
[105]. Staniszewska A D, Armenia J, King M, et al. PARP inhibition is a modulator of anti-tumor immune response in BRCA-deficient tumors[J]. Oncoimmunology, 2022, 11(1): 2083755.
[106]. Pantelidou C, Sonzogni O, De Oliveria Taveira M, et al. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer[J]. Cancer discovery, 2019, 9(6): 722-737.
[107]. Pantelidou C, Sonzogni O, De Oliveria Taveira M, et al. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer[J]. Cancer discovery, 2019, 9(6): 722-737.
[108]. Lampert E J, Zimmer A, Padget M, et al. Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: a proof-of-concept phase II study[J]. Clinical Cancer Research, 2020, 26(16): 4268-4279.
[109]. Jiao S, Xia W, Yamaguchi H, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression[J]. Clinical Cancer Research, 2017, 23(14): 3711-3720.
[110]. Powell J D, Pollizzi K N, Heikamp E B, et al. Regulation of immune responses by mTOR[J]. Annual review of immunology, 2012, 30(1): 39-68.
[111]. Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR[J]. Nature Reviews Immunology, 2015, 15(10): 599-614.
[112]. Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer[J]. New England Journal of Medicine, 2012, 366(6): 520-529.
[113]. Hare S H, Harvey A J. mTOR function and therapeutic targeting in breast cancer[J]. American journal of cancer research, 2017, 7(3): 383.
[114]. Witzig T E, Reeder C, Han J J, et al. The mTORC1 inhibitor everolimus has antitumor activity in vitro and produces tumor responses in patients with relapsed T-cell lymphoma[J]. Blood, The Journal of the American Society of Hematology, 2015, 126(3): 328-335.