







1. Introduction
Tuberculosis is a serious disease caused by Mycobacterium tuberculosis (MTB). It is estimated that approximately 10 million people worldwide are infected with MTB each year, and 1.5 million people die from the disease [1].
In some cases, M.tuberculosis can be eliminated by the immune system. However, in the other cases, even though M.tuberculosis are walled off, they remain alive and dormant. People with healthy immune system can generally resist M.tuberculosis. But people having compromised immune systems like HIV infection, malnutrition or diabetes may be affected easily.
M.tuberculosis is usually transmitted via inhalation, and commonly infects the lungs. M.tuberculosis usually involves particularly the right middle lobe and the right lower lobe of the lung tissue, which leads to the immune reaction. After that, macrophages will find the foreign cells then digest and destroy them. In most cases, after the macrophages engulfing foreign cells into phagosomes, those phagosomes will be fused with lysosomes that can almost break down any cells. However, the M.tuberculosis inhibits the lysosomes of the phagosomes so that MTB can survive. M.tuberculosis then starts to multiply within multiple macrophages of immune systems.
Approximately three weeks post-infection, the initial infection cells give rise to a granuloma, a structure formed by the immune system that aims to contain MTB and inhibits its dissemination. Within the central region of the granuloma, caseous necrosis develops, which is commonly referred to as the Ghon focus. The tissue encased by the granuloma typically experiences fibrosis with calcification.
2. Accurate structure prediction of biomolecular interactions with AlphaFold 3
Understanding the structure of proteins is critical for understanding biological mechanisms and designing effective drugs. AlphaFold, a practical tool, which can predicts the 3D structures of proteins based on their amino acid sequences. The recent development of alphafold3 offers high-accuracy prediction of complexes containing nearly all molecular types presenting in the Protein Data Bank [2]. The version's utility is unimaginable in the field of drug discoveries [3].
3. β-Lactamase BlaC and its Role in Antibiotic Resistance of MTB
β-lactamases are enzymes that allow MTB to withstand β-lactam antibiotics, a major category of antimicrobial medications. BlaC, a class A β-lactamase present in M. tuberculosis, plays a vital role in neutralizing antibiotics by dismantling the β-lactam ring structure.
Unlike some other β-lactamases, BlaC can act on a variety of β-lactams, such as penicillins, cephalosporins, and certain carbapenems, allowing M.tuberculosis to develop antibiotic resistance. For example, the resistance to the penicillin G. Penicillin G, contains a β-lactam ring. This four-membered ring is essential for t he antibiotic’s activity
When bacteria grow, they naturally produce autolysin, which is a kind of enzyme that break existing peptidoglycan bonds to allow for expansion. Normally, new polypetidoglycan strands can form cross-link quickly to maintain the strength for bacterial cell wall. So Penicillin kills bacteria by disrupting the synthesis of cross-link of polypetidoglycan cell walls. Since bacteria live in watery environments, water constantly enters the cell by osmosis. Without a sturdy cell wall to resist internal pressure, the bacteria swell and lyse ,which ultimately causing the bacterial cells to burst.
However, when penicillin G encounters BlaC, the enzyme cleaves the β-lactam ring, thus deactivating the antibiotic and allowing the bacterium to survive and proliferate. To overcome resistance, β-lactamase inhibitors have been developed to block the activity of enzymes like BlaC. These inhibitors, such as clavulanic acid and sulbactam, mimic the structure of β-lactam antibiotics and bind irreversibly to the active site of β-lactamases, preventing them from hydrolyzing the actual antibiotics.
Fortunately, the activity of blaC can be inhibited by β-lactamase inhibitors like clavulanate (CLA), which attaches to the enzyme's active site and stops it from hydrolyzing antibiotics.
4. ß-lactamase and drug resistant
According to their structure and function of the β-lactamases, they are categorized into several groups, including classes A, B, C, and D. These enzymes can hydrolyze various antibiotics to inactivate them; however, they can also be inhibited by different inhibitors.
Class A β-lactamases break down penicillins, first-generation cephalosporins, and aztreonam, which are usually inhibited by β-lactamase inhibitors such as clavulanic acid. Class B β-lactamases, which require zinc ions for catalysis, hydrolyze a broad range of β-lactams, including the carbapenems. However, Class B β-lactamases are resistant to β-lactamase inhibitors. Class C β-lactamases break down mainly the cephalosporins and are resistant to usual β-lactamase inhibitors. Class D β-lactamases break down oxacillin and, occasionally, the carbapenems.
In Mycobacterium tuberculosis (MTB), the broad substrate promiscuity of the β-lactamase BlaC compromines the efficacy of β-lactam antibiotics. This enzyme hydrolyzes the β-lactam ring catalytically, enabling the bacterium to resist the bactericidal effects of these drugs
5. Rv2068c blaC class A ß-lactamase in Mycobacterium tuberculosis
In M.tuberculosis, β-lactamase enzymes contribute to the bacteria's resistance to β-lactam antibiotics.
The principal β-lactamase enzyme of M.tuberculosis is BlaC. BlaC is class A β-lactamase and can catalyze a wide range of β-lactam antibiotics. BlaC has broad substrate specificity such that it is capable of inactivating, e.g., penicillins these antibiotics and the first-generation cephalosporins.
BlaC in M. tuberculosis functions by hydrolyzing the β-lactam antibiotic ring, thereby rendering the antibiotics inactive. This renders the antibiotics inactive so that they cannot inhibit cell wall synthesis in bacteria, allowing them to survive.
When meropenem is combined with clavulanate, it is a highly effective treatment process. The carbapenem (meropenem) is paired with the β-lactamase inhibitor clavulanate to effectively inhibit BlaC, allowing the β-lactam antibiotic to effectively target and kill the bacteria.
6. Results:Structural Comparisons of BlaC and Related Enzymes
Rv2068c BlaC, the primary β-lactamase in Mtb, is a 307-amino-acid protein with 18 alpha-helices and 9 beta-sheets. Comparative analyses with other β-lactamases reveal insights into structural variations and functional similaritie, here we use EMBL-EBI to compare 4 different β-lactamases. As shown in Figure 1[4]:
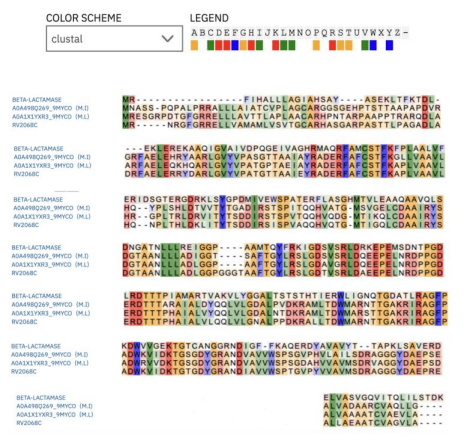
Rows: A row represents an individual β-lactamase sequence, designated by specific accession numbers.
Columns: Each column represents a position in the protein sequence where the amino acids from the various sequences are aligned.
Color Coding: Employed to emphasize conserved amino acids, facilitating the recognition of functionally significant areas.
In Figure 1, A0A1X1YXR3_9MYCO is a 308-amino-acid protein in Mycobacterium lacus, and A0A498Q269_9MYCO is a 307-amino-acid protein in Mycobacterium innocens. Both are hydrolases involved in antibiotic resistance mechanisms. GES-5, a beta-lactamase, is a 307-amino-acid protein specific to Klebsiella pneumoniae. It belongs to the Class A carbapenemase family and is a type of beta-lactamase. The Class A carbapenemases are common in having a disulfide bond between invariant residues Cys69 and Cys238, which is structurally stabilizing for these enzymes [5]. However, this disulfide bridge does not directly affect their antibiotic resistance activity. GES-5 provides resistance to a variety of antibiotics, including penicillins, cephalosporins, and carbapenems, and exhibits carbapenem-hydrolyzing activity.
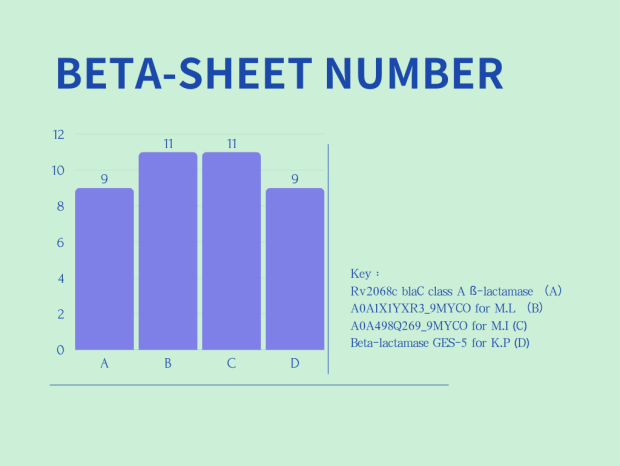
Comparisons of secondary structure of the 4 different β-lactamase are shown in Figure 2 :
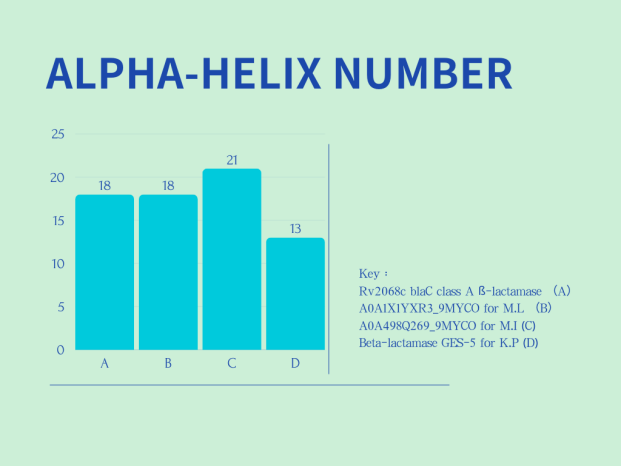
Left Panel (ALPHA-HELIX COUNT): The x-axis displays four distinct beta-lactamases: Rv2068c blaC class A β-lactamase (A), A0A1X1YXR3_9MYCO for M.L. (B), A0A498Q269_9MYCO for M.I. (C), and Beta-lactamase GES-5 for K.P (D). The y-axis shows the count of alpha-helices. The data indicates that C (A0A498Q269_9MYCO for M.I.) has the most alpha-helices, totaling 21. This is followed by B (A0A1X1YXR3_9MYCO for M.L.) and A (Rv2068c blaC class A β-lactamase), both having 18. D (Beta-lactamase GES-5 for K.P) has the least, with only 13 alpha-helices.
Right Panel (BETA-SHEET COUNT): The x-axis again represents the four different beta-lactamases, while the y-axis indicates the number of beta-sheets. In this chart, both B (A0A1X1YXR3_9MYCO for M.L.) and C (A0A498Q269_9MYCO for M.I.) have the highest count of beta-sheets, with 11 each. A (Rv2068c blaC class A β-lactamase) and D (Beta-lactamase GES-5 for K.P) each have 9 beta-sheets.
In summary, two charts in Figure 2 show the differences in the number of alpha-helices and beta-sheets among various β-lactamases, which are related to their structural stability and functionality.
7. Docking calculation for drug prediction against BlaC
Molecular docking is a computational method to predict the pose of the small molecule ligand when it binds to a large molecule(i.e. protein). Docking simulates the interaction between two molecules to seek the most energetically favorable binding orientations. Docking provides information about the ligand-receptor binding mode and affinity, thus facilitating the design of potential drugs.
A lower, more negative docking score indicates tighter binding of protein and ligand. The most favorable ligand should always be among the top-ranking clusters, since a ligand with low docking scores in many clusters can be a sign of tighter binding and binding affinity.
The docking results of 4 different potential ligands for Rv2068c blaC class A ß-lactamase using SwissDock are as shown in Table 1 [6][7][8]:
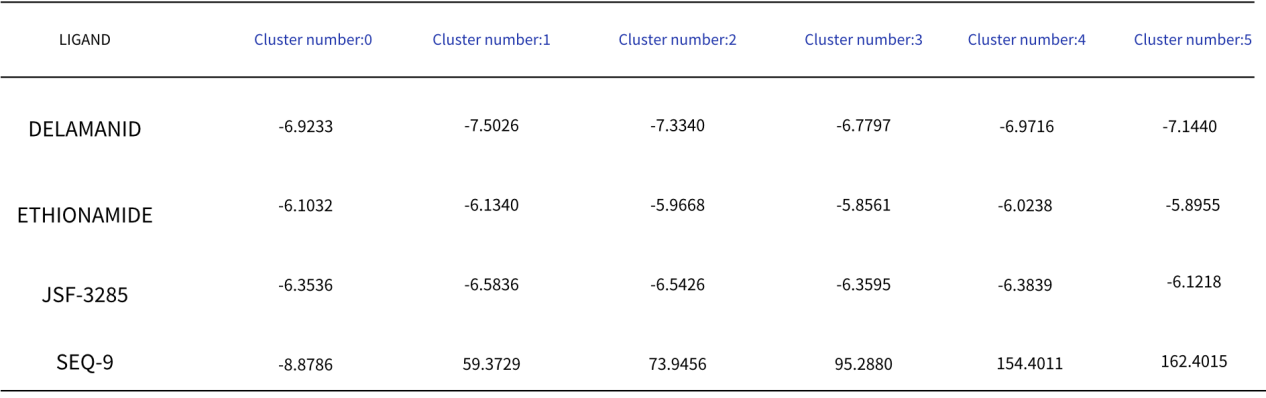
SWISSPARAM SCORE refers to the scoring metric used to evaluate how well each ligand binds to the protein. In molecular docking, a lower (more negative) score typically indicates a higher binding affinity.
LIGAND column lists the names of the ligands being tested. In this chart, there are four ligands: DELAMANID, ETHIONAMIDE, JSF-3285, and SEQ-9.
Cluster number represent different clusters or groups into which the docking results have been categorized. Each cluster number corresponds to a set of docking simulations or a different configuration of the protein-ligand complex.
Lower scores (more negative) suggest stronger binding.
In analyzing the docking results from Table 1, it was observed that DELAMANID exhibited relatively low and consistent docking scores across all clusters, suggesting a stable interaction with the protein. ETHIONAMIDE showed the lowest scores in clusters 3 and 5, indicating the strongest binding affinity in these specific clusters. JSF-3285 also demonstrated low and stable docking scores across all clusters, pointing to a consistent binding interaction. Conversely, SEQ-9 had the lowest score in cluster 0, but its scores were significantly higher in other clusters, suggesting weak binding interactions. Therefore, DELAMANID and JSF-3285 may be more suitable choices due to their consistently low docking scores across all clusters, which imply stable binding.
8. Discussion
8.1. Advantages of this Research
This research investigates how β-lactamase in MTB can resist various drugs by using AlphaFold 3 to predict the shape of the protein. This can help us know how the β-lactamase works and how it might be inhibited. Understanding this can help us know why some drugs of MTB do not work against tuberculosis infections. Also, while comparing various β-lactamases with others, this study illustrates the similarities and differences between various drug-resistant β-lactamases. Such information could prove to be beneficial while creating drugs to combat these resistant bacteria.
Potential Applications of This Study:
Understanding how drug-resistant MTB β-lactamases will help to come up with new medicines , which will be able to bypass drug-resistance of MTB, and it can improve existing treatments to make them even more potent, so they can work more effectively on drug-resistant infection. Further, understanding these β-lactamases more intimately may help design treatments specifically for MTB without damaging healthy bacteria.
8.2. Potential Limitations of This Study
While my research provides some findings, it is not exhaustive in addressing all aspects of drug resistance of MTB. More research is still required to understand this complex phenomenon. Second, the efficacy of β-lactamase in my research may not be identical to its efficacy within a living organism. That is, what works in my research may not work in the same manner in real infections.
9. Methods
To acquire the protein sequences necessary for my research, this research began with the use of UniProt to identify the Rv2068c blaC class A β-lactamase, a drug-resistant protein from MTB [9][10]. Following this, other β-lactamase proteins are found from different strains of tuberculosis that have similar sequence lengths to Rv2068c to ensure comparability in the analysis. By searching on UniProt, hree additional proteins are found: GES-5, A0A1X1YXR3, and A0A498Q269. These proteins, along with Rv2068c, provided a comprehensive datasets for this study on β-lactamase in tuberculosis
Protein sequences obtained from UniProt are copied to input protein sequences into AlphaFold3. AlphaFold3 predicted the protein structures, allowing visualization of their 3D spatial configurations.
Afterward, structural comparisons of the four β-lactamase proteins—Rv2068c, blaC class A β-lactamase, Beta-lactamase GES-5, A0A1X1YXR3_9MYCO, and A0A498Q269_9MYCO—were conducted using a combination of amino acid sequence alignment and secondary structure analysis. The alignment of sequences can detect conserved domains, which are critical to comprehend the similarities and disparities in the functionality of these proteins. In addition, sequence alignment can identify conserved domains, which are essential for understanding functional similarities and differences between proteins. Additionally, analyzing secondary structures—such as the number of alpha-helices and beta-sheets—reveals variations in their architectural features. Through this, it was possible to relate protein structural changes to their stability and functional potential, which can tell how these β-lactamases enable antibiotic resistance mechanisms.
To do the docking, molecular structure files are needed by first downloading the 3D structure of Rv2068c blaC class A β-lactamase from the AlphaFold Protein Structure Database in PDB format [11][12]. Suitable ligands were then identified on guidetopharmacology.org, and then, their SMILES strings are copied [13][14]. The protein's 3D structure and the SMILES strings were input into SWISSDOCK to carry out the docking process. Following this, a comparism between the docking results to analyze,and summarize to identify those with the highest affinity for Rv2068c blaC class A β-lactamase.
References
[1]. World Health Organization. (2024). Tuberculosis. Retrieved from https://www. who. int/health-topics/tuberculosis#tab=tab_1
[2]. DeepMind. (2024). AlphaFold Server. Retrieved from https://alphafoldserver. com/welcome
[3]. Desai, D. , Kantliwala, S. V. , Vybhavi, J. , Ravi, R. , Patel, H. , & Patel, J. (2024). Review of AlphaFold 3: Transformative Advances in Drug Design and Therapeutics. Cureus, 16(7), e63646. https://doi. org/10. 7759/cureus. 63646
[4]. European Bioinformatics Institute. (2024). EMBL-EBI homepage. Retrieved from https://www. ebi. ac. uk/
[5]. Smith, C. A. , Nossoni, Z. , Toth, M. , Stewart, N. K. , Frase, H. , & Vakulenko, S. B. (2016). Role of the Conserved Disulfide Bridge in Class A Carbapenemases. The Journal of biological chemistry, 291(42), 22196–22206. https://doi. org/10. 1074/jbc. M116. 749648
[6]. Bugnon, M. , Röhrig, U. F. , Goullieux, M. , Perez, M. A. S. , Daina, A. , Michielin, O. , & Zoete, V. (2024). SwissDock 2024: major enhancements for small-molecule docking with Attracting Cavities and AutoDock Vina. Nucleic acids research, 52(W1), W324–W332. https://doi. org/10. 1093/nar/gkae300
[7]. Grosdidier, A. , Zoete, V. , & Michielin, O. (2011). SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic acids research, 39(Web Server issue), W270–W277. https://doi. org/10. 1093/nar/gkr366
[8]. Swiss Institute of Bioinformatics. (2024). SwissDock. Retrieved from https://www. swissdock. ch/
[9]. UniProt Consortium (2025). UniProt: the Universal Protein Knowledgebase in 2025. Nucleic acids research, 53(D1), D609–D617. https://doi. org/10. 1093/nar/gkae1010
[10]. UniProt. (2024). Retrieved from https://www. uniprot. org/
[11]. Jumper, J. , Evans, R. , Pritzel, A. , Green, T. , Figurnov, M. , Ronneberger, O. , Tunyasuvunakool, K. , Bates, R. , Žídek, A. , Potapenko, A. , Bridgland, A. , Meyer, C. , Kohl, S. A. A. , Ballard, A. J. , Cowie, A. , Romera-Paredes, B. , Nikolov, S. , Jain, R. , Adler, J. , Back, T. , … Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi. org/10. 1038/s41586-021-03819-2
[12]. AlphaFold Protein Structure Database. (2024). Retrieved from https://alphafold. ebi. ac. uk/
[13]. Harding, S. D. , Armstrong, J. F. , Faccenda, E. , Southan, C. , Alexander, S. P. H. , Davenport, A. P. , Spedding, M. , & Davies, J. A. (2024). The IUPHAR/BPS Guide to PHARMACOLOGY in 2024. Nucleic acids research, 52(D1), D1438–D1449. https://doi. org/10. 1093/nar/gkad944
[14]. Guide to Pharmacology. (2024). Retrieved from https://www. guidetopharmacology. org/
Cite this article
Gong,L. (2025). Prediction of Protein Structure of β-LactamaseEnzymes in Mycobacterium Tuberculosis and Drug Prediction Based on AlphaFold3. Theoretical and Natural Science,119,25-31.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of ICEGEE 2025 Symposium: Sensor Technology and Multimodal Data Analysis
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. World Health Organization. (2024). Tuberculosis. Retrieved from https://www. who. int/health-topics/tuberculosis#tab=tab_1
[2]. DeepMind. (2024). AlphaFold Server. Retrieved from https://alphafoldserver. com/welcome
[3]. Desai, D. , Kantliwala, S. V. , Vybhavi, J. , Ravi, R. , Patel, H. , & Patel, J. (2024). Review of AlphaFold 3: Transformative Advances in Drug Design and Therapeutics. Cureus, 16(7), e63646. https://doi. org/10. 7759/cureus. 63646
[4]. European Bioinformatics Institute. (2024). EMBL-EBI homepage. Retrieved from https://www. ebi. ac. uk/
[5]. Smith, C. A. , Nossoni, Z. , Toth, M. , Stewart, N. K. , Frase, H. , & Vakulenko, S. B. (2016). Role of the Conserved Disulfide Bridge in Class A Carbapenemases. The Journal of biological chemistry, 291(42), 22196–22206. https://doi. org/10. 1074/jbc. M116. 749648
[6]. Bugnon, M. , Röhrig, U. F. , Goullieux, M. , Perez, M. A. S. , Daina, A. , Michielin, O. , & Zoete, V. (2024). SwissDock 2024: major enhancements for small-molecule docking with Attracting Cavities and AutoDock Vina. Nucleic acids research, 52(W1), W324–W332. https://doi. org/10. 1093/nar/gkae300
[7]. Grosdidier, A. , Zoete, V. , & Michielin, O. (2011). SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic acids research, 39(Web Server issue), W270–W277. https://doi. org/10. 1093/nar/gkr366
[8]. Swiss Institute of Bioinformatics. (2024). SwissDock. Retrieved from https://www. swissdock. ch/
[9]. UniProt Consortium (2025). UniProt: the Universal Protein Knowledgebase in 2025. Nucleic acids research, 53(D1), D609–D617. https://doi. org/10. 1093/nar/gkae1010
[10]. UniProt. (2024). Retrieved from https://www. uniprot. org/
[11]. Jumper, J. , Evans, R. , Pritzel, A. , Green, T. , Figurnov, M. , Ronneberger, O. , Tunyasuvunakool, K. , Bates, R. , Žídek, A. , Potapenko, A. , Bridgland, A. , Meyer, C. , Kohl, S. A. A. , Ballard, A. J. , Cowie, A. , Romera-Paredes, B. , Nikolov, S. , Jain, R. , Adler, J. , Back, T. , … Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi. org/10. 1038/s41586-021-03819-2
[12]. AlphaFold Protein Structure Database. (2024). Retrieved from https://alphafold. ebi. ac. uk/
[13]. Harding, S. D. , Armstrong, J. F. , Faccenda, E. , Southan, C. , Alexander, S. P. H. , Davenport, A. P. , Spedding, M. , & Davies, J. A. (2024). The IUPHAR/BPS Guide to PHARMACOLOGY in 2024. Nucleic acids research, 52(D1), D1438–D1449. https://doi. org/10. 1093/nar/gkad944
[14]. Guide to Pharmacology. (2024). Retrieved from https://www. guidetopharmacology. org/