







1. Introduction
Alzheimer's disease (AD) is a central nervous system degenerative condition that typically affects elderly people and is characterized by gradual cognitive dysfunction and behavioral impairment. As the average human lifetime rises, AD is becoming more and more common. β-amyloid deposition is currently the most influential hypothesis in the study of the pathogenesis of AD. In 2021, when investigating Simufilam, an experimental drug for AD, Matthew Schrag found that a Nature article published in 2006 with Lesné as the first author and Karen Ashe, a renowned neuroscientist and professor at the University of Minnesota (UMN), as the corresponding author, appeared to contain multiple doctored images [1]. At that time, the attack on the Aβ deposition hypothesis was overwhelming and many neurobiologists began to question the Aβ deposition hypothesis. This review paper describes the development of β-amyloid precipitation hypothesis and introduces what Aβ42 and Aβ*56 are. Besides, the effects of Aβ42 and Aβ*56 on the human body is also analyzed. This paper helps prove that β-amyloid precipitation can be studied further.
2. The development of β-amyloid precipitation hypothesis
Memory loss, frequent lying to cover up negligent behavior, confusion, agitation and anxiety, and mania are typical symptoms of AD. Originally, scientists discovered through autopsy that patients with AD had reduced brain size, tangled neurogenic fibers, and degenerated neuronal cells [2]. To this day, scientists have named the deposits in the brains of Alzheimer's patients beta-amyloid, and they think that the deposition of β amyloid (Aβ) is one possible biomarker for Alzheimer. With the development of many novel radiotracers that bind to fibrillar Aβ, sensitive estimations of amyloid deposition in diverse brain areas are now possible [3].
According to the author of the article "A particular amyloid-protein assembly in the brain impairs memory" published in 2006, when given to young rats, A*56 isolated from the brains of damaged Tg2576 mice damages memory. A*56 affects memory in a way that is unrelated to plaque formation or neuronal death and may be a factor in the cognitive deficits associated with AD [4]. Cited more than 2,200 times, this article is the seminal work in the field of the "amyloid" hypothesis of AD (Figure 1). What Figure 2 is trying to convey is that, as the mice in the AD model get older, the expression level of the Aβ*56 protein increases [1]. In fact, the falsification may not mean that the "amyloid" hypothesis is wrong since only the Aβ*56 protein is suspected to be falsified, and there are many other Aβ oligomers that have been shown to be neurotoxic. The group of Sylvain Lesné points out that Aβ*56 is the culprit [4]. Steven and Qin put forward that Aβ42 will cause Alzheimer’s disease [5,6]. The following sections mainly argue about these views.
Figure 1. Number of citations of articles that found to be image falsified [4].

Figure 2. The data Ashe provided on PubPeer after Schrag challenged the original paper's data, which still shows signs of copying [1].
3. Previous studies on Aβ*56
3.1. How Aβ*56 was found to be damaging to memory
The decline in memory function that occurs with aging is thought to be caused by changes in synaptic function. It is also believed that some individuals with AD develop the disease through neurodegeneration. To study the causes of this loss, Sylvain Lesné and other biologists used a mouse that has a variant of the human amyloid precursor protein. The young Tg2576 mice had normal memory, while the older ones had neuritis plaques that were filled with amyloid-β. Middle-aged Tg2576 animals, on the other hand, developed memory deficits without loss of neurons. Sylvain Lesné and other biologists discovered that the deficits in these animals were caused by the accumulation of 56-kDa of soluble amyloid-β in the extracellular matrix. In the brains of damaged Tg2576 mice, Aβ*56 was purified. When purified Aβ*56 was given to young rats, the animals exhibited memory deficits. Therefore, scientists are believed that this substance can contribute to the development of AD [4].
3.2. The effect of Aβ*56
It is thought that the primary beginning component in neurodegenerative illnesses, such as AD, is the production of oligomers of amyloid-forming proteins. These have been linked to the phosphorylation and aggregation of tau. From the AD model Tg2576 mice, several Aβ variants were identified. Amar and more researchers found that modifications in neuronal signaling were induced by the 56-kDa oligomer A*56, which is known to be detected prior to the onset of illness symptoms in patients. A*56 interacts with N-methyl-d-aspartate receptors in primary cortical neurons. This interaction increases the Ca2+ influx and the concentration of Ca2+ in the cytoplasm. It also activates the CaMKII protein, which is known to be involved in the development of AD. In Tg2576 mice, the activation of this protein was linked to an increase in the site-specific phosphorylation and the mis sorting of tau. In contrast, the effects of exposure to other oligomers, such as trimers and dimers, were not detected in cultured primary cortical neurons. These findings suggest that the various pathways that are involved in the activation of neurons can be efficiently targeted by the different Aβ assemblies [7].
4. Previous studies on Aβ42
4.1. The formation of Aβ 42
Studies have shown that the amyloid β-protein, which is a component of AD, plays a central role in the development of the disease. As shown in Figure 3, it is produced by two aspartic enzymes, β- and γ-secretases. The former cleaves APP and allows it to be shed into the extracellular fluid and luminal. The latter leaves a C-terminal residue. After the amino acid long stub is cleaved by γ-secretase, the resulting Aβ is released [8].
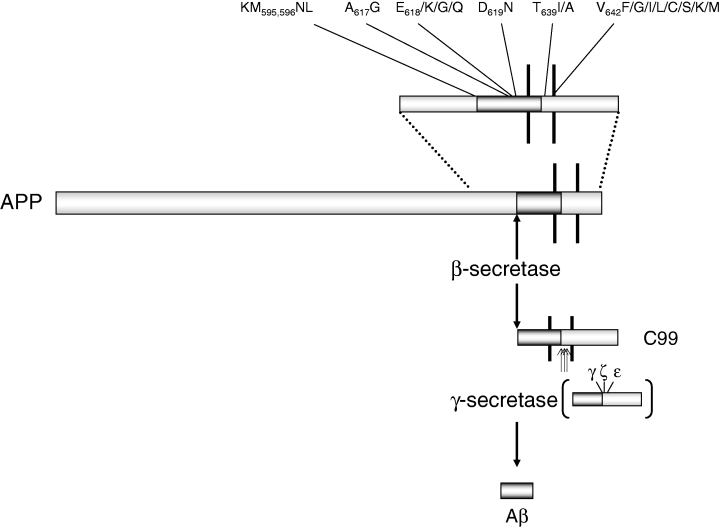
Figure 3. β-Amyloid precursor protein (APP) mutations and processing [8].
Depending on the point at which the cleavage occurs, three different forms of Aβ are produced. These include 38, 40, and 42 amino acid residues. The relative quantity of A42 formed is noteworthy because it is more prone to forming amyloid fibrils and oligomerizing than the more abundantly produced A40 peptide [8].
4.2. The effect of Aβ42
Although it is known that the accumulation of Aβ can lead to AD, it is also believed that the protein can function as a neurotrophic factor. Some studies suggest that it can stimulate the growth of neurons and promote the development of antimicrobial peptides in the innate immune system. Meanwhile, Aβ could potentially be involved in the activation of phosphokinase and the transportation of cholesterol [5].
According to the amyloid hypothesis, Aβ monomer is not harmful. After aggregation, it can cause harmful effects, though. These include heightened membrane permeability, elevated oxidative stress, altered cell skeleton, apoptotic pathway activation, and memory retention deficits. Blood, brain, and CSF samples containing stable low-molecular-weight A-42 oligomers were found to be related to the prevalence of AD [5].
A basic consequence of mutations linked to familial Alzheimer's disease (FAD) is an increase in Aβ42 extracellular concentration. Aβ42 is deposited early and selectively in senile plaques, which are an unchanging hallmark of all forms of AD, suggesting that this action of FAD-associated mutations may be directly related to the pathophysiology of the disease. The findings of Steven support the idea that brain Aβ deposition is a crucial early event in the etiology of all forms of AD and provides strong evidence that FAD-associated mutations all contribute to Aβ deposition by raising the extracellular concentration of Aβ42 [6].
5. The difference between Aβ42 and Aβ40
The most significant Aβ isomers are seen as being Aβ40 and Aβ42. The only distinction between them is an extra isoleucine and an alanine at the C-terminus of Aβ42 [5].
Because of its link to toxicity caused by the Aβ peptide, copper is recognized as a significant contributor to the pathophysiology of AD. According to Lu Jin's research, copper changes the morphology and structure of A-42 and increases its toxicity, while it has no discernible impact on A-40. They draw the conclusion that copper induces the development of A-42 nanoscale oligomers, which is a crucial process in neurotoxicity [9].
According to the research of Chang’s team, a high Aβ40/Aβ42 ratio shields neurons against the harmful effects of Aβ42. This may imply that restoring the proper ratio of Aβ. The potential synergistic interactions between βA42 and Aβ40 in vivo, however, are not well understood [10]. Kim J’s group also found that Aβ40 has a strong anti-amyloidogenic effect in the brain of Tg2576 mice and is protective during pathogenesis [11].
6. Diagnosing Alzheimer's disease
The increasing importance of the diagnosis and validation of AD and other forms of dementia has led to the development of new diagnostic tools. Currently, the most common method for determining the presence of β-amyloid (1–42), total tau and phospho-tau-181 in cerebrospinal fluid (CSF) is the ELISA [12].
Hansson, O ‘s group’s finding also suggest that when analyzing CSF AD biomarkers, the CSF Aβ42/40 ratio should be used rather than the absolute value of CSF Aβ42 to increase the proportion of patients correctly diagnosed [13].
7. Conclusion
Although a seminal article on AD was revealed to be a falsification of images, the β-amyloid hypothesis cannot be completely rejected. In addition to Aβ*56, there are various oligomers of beta amyloid, including Aβ42 and Aβ40. These other oligomers of beta amyloid have been suggested by other scientists to have more or less toxic effects on the human brain. This paper also aims to use some previous studies to show that this hypothesis should not be completely abandoned, but more funds and manpower should be devoted to the study of other β-amyloid oligomers. The pathogenesis of AD is still a mystery, so, any research direction should not be given up.
References
[1]. Piller, C. Blots on a field?. Science (New York, N.Y.) 377(6604), 358–363 (2022). https://doi.org/10.1126/science.add9993.
[2]. Gurwitz, D. Auguste D and Alzheimer's disease. Lancet (London, England) 350(9073), 298 (1997). https://doi.org/10.1016/S0140-6736(05)62274-X.
[3]. Rodrigue, K.M., Kennedy, K.M., Park, D.C. Beta-Amyloid Deposition and the Aging Brain. Neuropsychol Rev 19, 436 (2009). https://doi.org/10.1007/s11065-009-9118-x.
[4]. Lesné, S., Koh, M., Kotilinek, L. et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 (2006). https://doi.org/10.1038/nature04533.
[5]. Qiu, T., Liu, Q., Chen, Y.X., Zhao, Y.F., Li, Y.M. Aβ42 and Aβ40: similarities and differences. J Pept Sci 21(7), 522-9 (2015). doi: 10.1002/psc.2789. Epub 2015 May 28. PMID: 26018760.
[6]. Steven, G. Younkin, The role of Aβ42 in Alzheimer's disease. Journal of Physiology-Paris 92(3–4), 289-292 (1998). https://doi.org/10.1016/S0928-4257(98)80035-1.
[7]. Amar, F., Sherman, M.A., Rush, T., Larson, M., Boyle, G., Chang, L., Götz, J., Buisson, A., Lesné, S.E. The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci Signal 10(478), eaal2021 (2017). doi: 10.1126/scisignal.aal2021. PMID: 28487416; PMCID: PMC5859319.
[8]. Walsh, D.M., Selkoe, D.J. Aβ Oligomers – a decade of discovery. Journal of Neurochemistry 101, 1172-1184 (2007). https://doi.org/10.1111/j.14714159.2006.04426.x.
[9]. Jin, L., Wu, W.H., Li, Q.Y., Zhao, Y.F., Li, Y.M. Copper inducing Aβ42 rather than Aβ40 nanoscale oligomer formation is the key process for Aβneurotoxicity. Nanoscale 3, 4746–4751 (2011).
[10]. Chang, C.C., Althaus, J.C., Carruthers, C.J., Sutton, M.A., Steel, D.G., Gafni, A. Synergistic interactions between Alzheimer’s Aβ40 and Aβ42 on the surface of primary neurons revealed by single molecule microscopy. PLoS One 8, e82139 (2013).
[11]. Kim, J., Onstead, L., Randle, S., Price, R., Smithson, L., Zwizinski, C., Dickson, D.W., Golde, T., McGowan, E. Aβ40 inhibits amyloid deposition in vivo. J. Neurosci 27, 627–633 (2007).
[12]. Humpel, C. Identifying and validating biomarkers for Alzheimer's disease. Trends in Biotechnology 29(1), 26-32 (2011). https://doi.org/10.1016/j.tibtech.2010.09.007.
[13]. Hansson, O., Lehmann, S., Otto, M. et al. Advantages and disadvantages of the use of the CSF Amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alz Res Therapy 11, 34 (2019). https://doi.org/10.1186/s13195-019-0485-0.
Cite this article
Li,B. (2023). A review on the β-amyloid precipitation hypothesis. Theoretical and Natural Science,6,45-50.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the International Conference on Modern Medicine and Global Health (ICMMGH 2023)
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Piller, C. Blots on a field?. Science (New York, N.Y.) 377(6604), 358–363 (2022). https://doi.org/10.1126/science.add9993.
[2]. Gurwitz, D. Auguste D and Alzheimer's disease. Lancet (London, England) 350(9073), 298 (1997). https://doi.org/10.1016/S0140-6736(05)62274-X.
[3]. Rodrigue, K.M., Kennedy, K.M., Park, D.C. Beta-Amyloid Deposition and the Aging Brain. Neuropsychol Rev 19, 436 (2009). https://doi.org/10.1007/s11065-009-9118-x.
[4]. Lesné, S., Koh, M., Kotilinek, L. et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 (2006). https://doi.org/10.1038/nature04533.
[5]. Qiu, T., Liu, Q., Chen, Y.X., Zhao, Y.F., Li, Y.M. Aβ42 and Aβ40: similarities and differences. J Pept Sci 21(7), 522-9 (2015). doi: 10.1002/psc.2789. Epub 2015 May 28. PMID: 26018760.
[6]. Steven, G. Younkin, The role of Aβ42 in Alzheimer's disease. Journal of Physiology-Paris 92(3–4), 289-292 (1998). https://doi.org/10.1016/S0928-4257(98)80035-1.
[7]. Amar, F., Sherman, M.A., Rush, T., Larson, M., Boyle, G., Chang, L., Götz, J., Buisson, A., Lesné, S.E. The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci Signal 10(478), eaal2021 (2017). doi: 10.1126/scisignal.aal2021. PMID: 28487416; PMCID: PMC5859319.
[8]. Walsh, D.M., Selkoe, D.J. Aβ Oligomers – a decade of discovery. Journal of Neurochemistry 101, 1172-1184 (2007). https://doi.org/10.1111/j.14714159.2006.04426.x.
[9]. Jin, L., Wu, W.H., Li, Q.Y., Zhao, Y.F., Li, Y.M. Copper inducing Aβ42 rather than Aβ40 nanoscale oligomer formation is the key process for Aβneurotoxicity. Nanoscale 3, 4746–4751 (2011).
[10]. Chang, C.C., Althaus, J.C., Carruthers, C.J., Sutton, M.A., Steel, D.G., Gafni, A. Synergistic interactions between Alzheimer’s Aβ40 and Aβ42 on the surface of primary neurons revealed by single molecule microscopy. PLoS One 8, e82139 (2013).
[11]. Kim, J., Onstead, L., Randle, S., Price, R., Smithson, L., Zwizinski, C., Dickson, D.W., Golde, T., McGowan, E. Aβ40 inhibits amyloid deposition in vivo. J. Neurosci 27, 627–633 (2007).
[12]. Humpel, C. Identifying and validating biomarkers for Alzheimer's disease. Trends in Biotechnology 29(1), 26-32 (2011). https://doi.org/10.1016/j.tibtech.2010.09.007.
[13]. Hansson, O., Lehmann, S., Otto, M. et al. Advantages and disadvantages of the use of the CSF Amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alz Res Therapy 11, 34 (2019). https://doi.org/10.1186/s13195-019-0485-0.