







1. Introduction
Acute myeloid leukemia (AML) has been known for its rapid growth of abnormal cells since its discovery, killing tens of thousands of people each year. In the past few decades of clinical treatment, salvage chemotheraphy has had little positive effect on patients with AML, which has also made the clinical treatment of AML more difficult. With the advancement of medical science in the 21st century, more and more targeted drugs are synthesized to treat various types of cancer, also, the use of targeted drugs to destroy cancer cells is considered to be one of the best ways to treat cancer today. The oral, powerful and selective second-generation FLT3 inhibitor gilteritinib, which exhibits single-agent effectiveness against relapsed or refractory FLT3-mutant AML, has steadily gained attention in recent years, as described in this work [1]. The US FDA authorized gilteritinib in November 2018 as a second-generation FLT3 inhibitor for the therapy of relapsed or resistant AML brought on by FLT3 gene mutations. Additionally, according to NCCN recommendations for 2019, it was the third FLT3-targeted medication for the therapy of refractory relapsed AML. Compared to first-generation drugs, gilteritinib has no off-target activity and is less effective in newly diagnosed patients, but it is also more targeted, meaning it can assist patients undergoing refractory or relapsed therapy more effectively and creates the possibility of combination therapy with first-generation inhibitors.
Although gilteritinib has been involved in the clinical treatment of AML, due to its short development time and fewer options for its synthesis routes, it still needs to be continuously developed and optimized. In this paper, it will start from the mechanism of action of gilteritinib and the target, analysis of its different synthetic routes, evaluation of their safety, efficiency, economy and other aspects. The discussion of these questions will be of great significance for the further industrial production of the drug and the future treatment of AML patients.
2. Mechanism of action of gilteritinib with FLT3
2.1. FLT3 mutation mechanism
An anti-tumor kinase inhibitor called gilteritinib is used to treat acute myelocytic leukemia (AML). Massive underlying cytogenetic, chromosomal anomalies can be seen in AML, a cancer. FLT3 is the most typical mutation. The FLT3 gene, which is located on chromosome 13q12 is transcribed under supervision of FLT3 transmembrane receptor tyrosine kinase (RTK). The FLT3 ligand (FL) then activates the FLT3 gene, which is expressed on certain bone marrow and lymphoid progenitor cells. The tyrosine kinase (TK), external, kinase insertion, paramembrane, and c-terminal intracellular domains of FLT3 are among its five domains, as seen in Figure 1. It is a component of multiple signaling pathways that control several aspects of the cell's life cycle, including differentiation and apoptosis. FL is created by bone marrow stromal cells, which is available in membrane-bound or soluble form. In general, RTK is dimerized, phosphorylated, and activated when FL attaches to the FLT3 extracellular domain. FLT3 is only activated by negative feedback when necessary since the concentration of soluble FL is typically quite low, but it can rise dramatically under undeveloped circumstances [2]. Internal tandem duplication (ITD) mutations and tyrosine kinase domain (TKD) mutations are the two different types of FLT3 mutations [3]. Both types of mutations cause abnormal changes in leukemia cell proliferation, differentiation, and survival [4, 5]. Gilteritinib can significantly reduce the phosphorylation level of MV4-11 and MOLM13 cell line FLT3 protein and downstream molecules STAT5, ERK and AKT protein, which increase 160KD FLT3 protein’s expression on the membrane surface of MV4-11, MOLM13 cells. In turn, leukemia cells can proliferate, differentiate and survive normally, thereby treating leukemia.
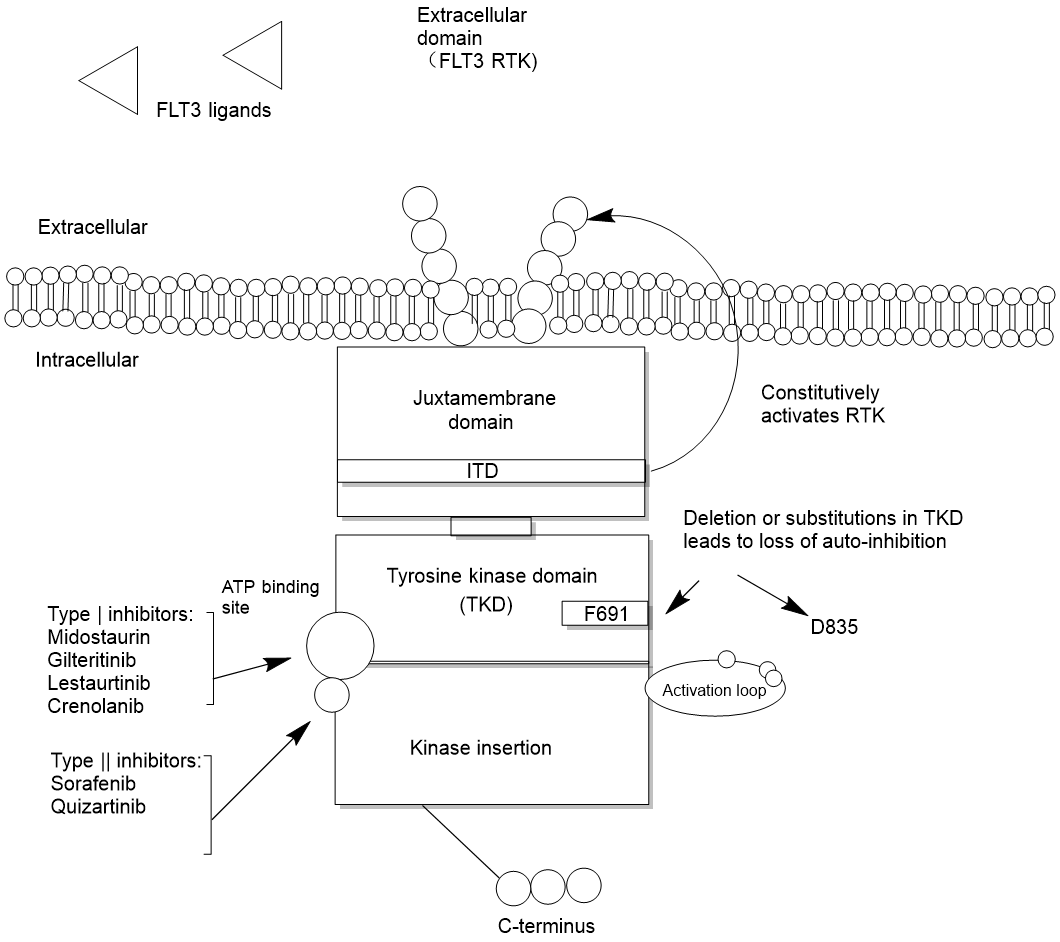
Figure 1. The structure and mechanism of FLT3.
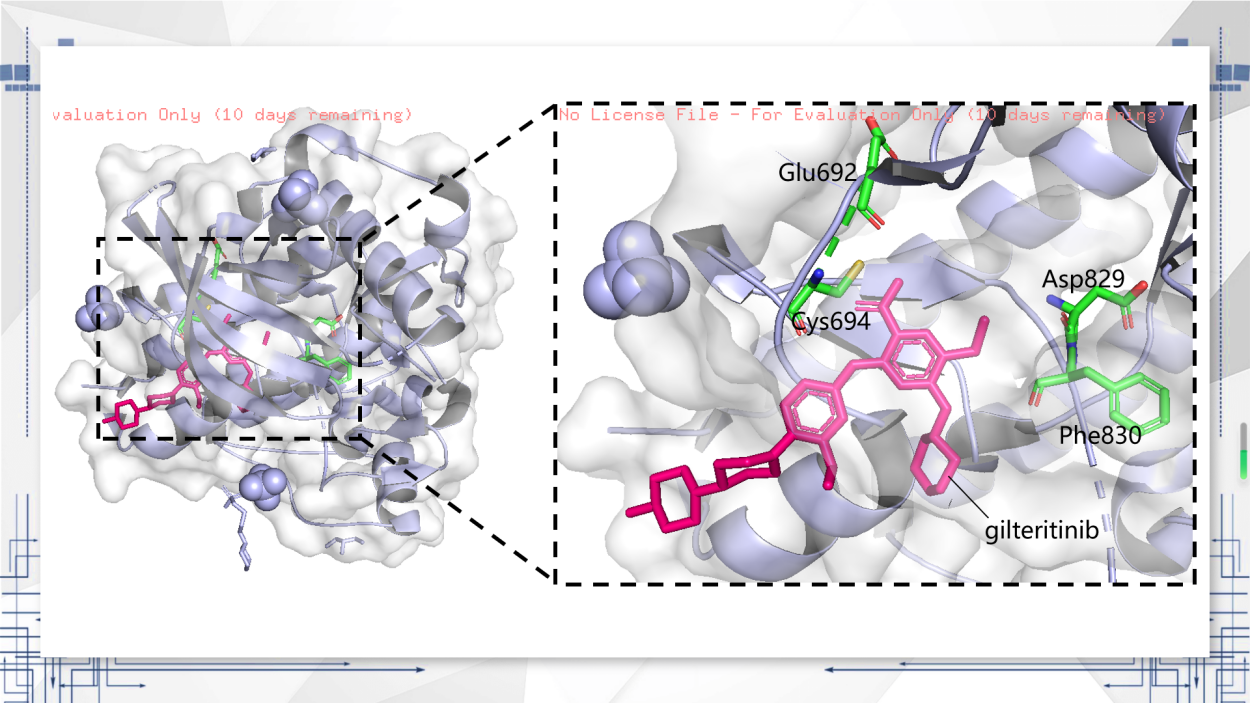
Figure 2. FLT3 crystal 3D model.
2.2. Gilteritinib mechanism of action
In order to see the binding pattern, as seen in Figure 2, the molecular model of gilteritinib with the FLT3 kinase domain compound was plotted. Gilteritinib binds to FLT3’s ATP pocket as predicted by computational modeling in the publication by Mori et al. (2017), assayed that Asp829 and Phe830 use the "DFG-out" conformation [6]. There is no significant relationship between gilteritinib and activation loops. For gilteritinib’s binding mode, the carbamoyl group has hydrogen bonds before the backbone atoms of Glu692, Cys694 [7].
Griffith et al. (2004) reported that the model shown between FLT3 and gilteritinib was synthesized by the FLT3wt’s kinase domain, which include the juxtamembrane (JM) domain, and that the active site of JM domain with FLT3 was able to bind and stabilize the auto-inhibitory form [8].
3. Gilteritinib synthetic route assessment
Currently, as a new drug, there are few synthetic routes for gilteritinib, and the synthesis process is not mature enough, there are three main synthetic routs will be evaluated below.
3.1. Synthetic route 1
As can be seen in Figure 3, route 1 is reported by the original patent, with ethyl propionylaecetate as the starting material, decompressed by nitrosation to obtain propanal, oxime and then through annulation, chlorination and hydrolysis to obtain 3,5-dichloro-6-ethylpyrazinecarboxamide as intermediate product, gilteritinib is prepared by condensation and nucleophilicsubstitution reaction [9].
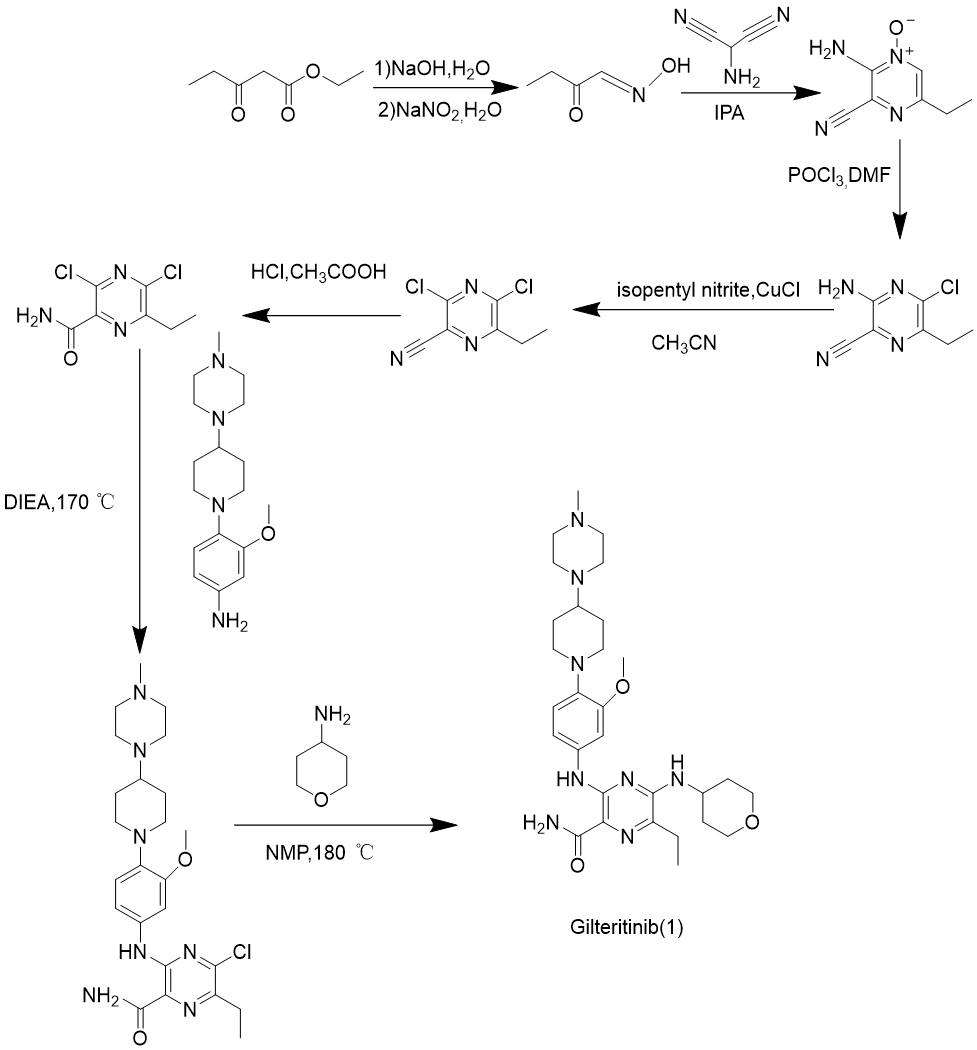
Figure 3. Gilteritinib synthesis route 1.
\( Atom economy=\frac{Molecular mass of desired}{Molecular mass of all reactants}×100\% \ \ \ (1) \)
The reaction conditions are harsh and require high temperatures throughout the process, which is relatively dangerous. The by-products are relatively simple and easy to handle, and the safety is poor. The target product can be obtained by sequentially replacing the two halogen atoms of the raw material. The electrophilicity of the carbon where the two halogen atoms are located is almost the same. Despite the microwave assisted nucleophilic substitution of the side chain, there are still side reactions that occur, resulting in low efficiency. A two-step substitution reaction can be obtained, but it requires microwave catalysis assistance and is not suitable for large-scale industrial production, with average convenience. The reaction is assisted by microwave, and the atomic utilization rate calculated according to equation (1) is relatively high, at 53.84% (In the case of not taking into account by-products). The by-products are simple, reach the green chemical standards, and have certain progressiveness.
3.2. Synthetic route 2
As is shown in Figure 4, route 2 uses the same raw materials and methods as route 1 to obtain propanal, oxime, and then passes an annulation reaction, chlorination, bromination to get intermediate product: 3-bromo-5-chloro-6-ethylpyrazine-2-carbonitrile, and gilteritinib is prepared by nucleophilic substitution, Buchwald coupling, and hydrolysis [10].
The reaction conditions of this synthesis route are relatively mild, but the organic toxic impurities generated by the side reactions are difficult to remove, which does not comply with green chemistry and has relatively average safety. It also replaces the two halogen atoms on the six membered ring of the raw material first, and then hydrolyzes the side chain groups on the ring to obtain the final product. Due to the small difference in the electrophilicity of the carbon where the two halogen atoms are located, side reactions such as side chain inversion are prone to occur, resulting in low efficiency. The final product can be obtained by simply replacing the halogen atoms on the six membered ring twice and then hydrolyzing it, which is more convenient. The theoretical atom utilization rate of this synthesis route is 50.75%. However, due to the poor reaction selectivity, there are a large number of toxic organic by-products that are difficult to remove, resulting in the actual atom utilization rate being lower than the theoretical value, which does not meet the green chemical standard, and does not have progressiveness.
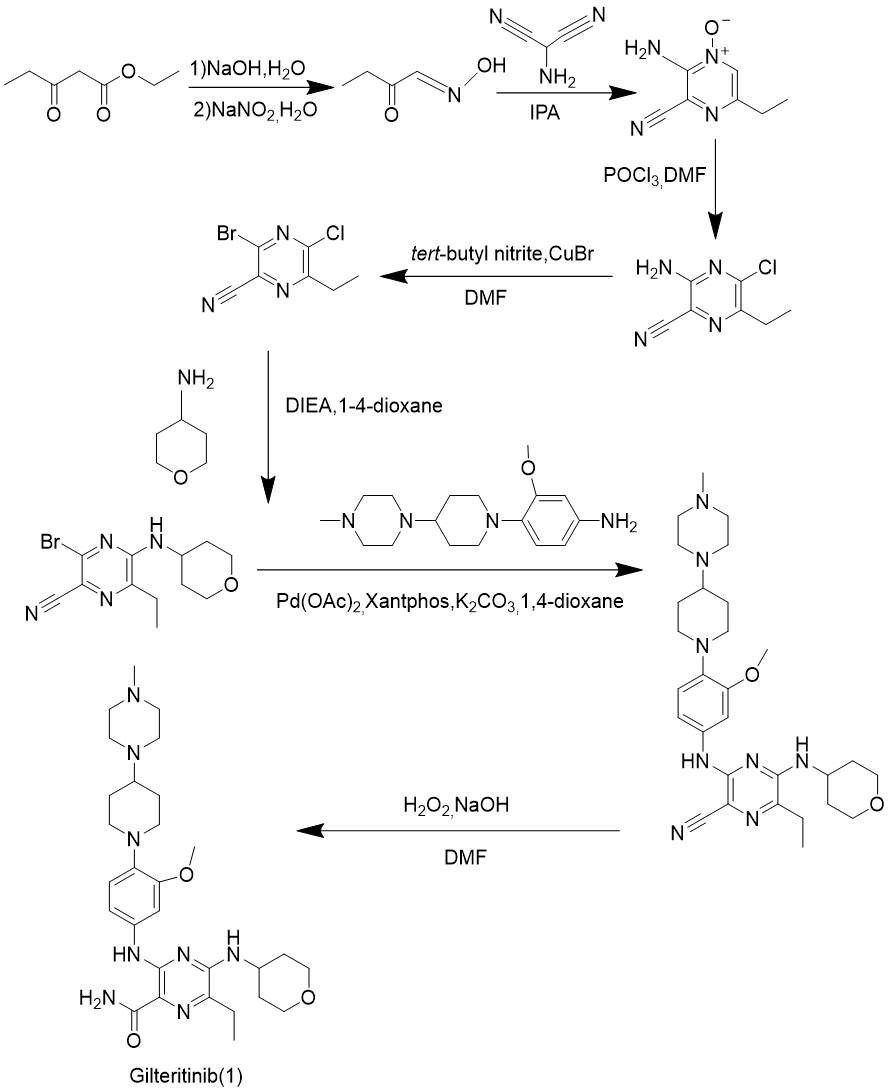
Figure 4. Gilteritinib synthesis route 2.
3.3. Synthetic route 3
Route 3 is the result of improvement of route 1 and route 2 through Li et al. (2021), which is shown in Figure 5 [10]. Route 3 gets 3-amino-5-chloro-6-methyl-pyrazine-2-carbonitrile (2) as the intermediate product by a method similar to that of route 1 and route 2, reacting with 4-aminotetrahydropyran (3) by nucleophilic substitution reaction to gain 3-amino-6-methyl-5- [(tetrahydro-2H-pyran-4-yl) amino]-2-carbonitrile (4), 3-methoxy-4- (4-(4-methylpiperidin-1-yl) piperidin-1-yl) aniline (5) as intermediate material, Sandmeyer reaction to generate chlorinated substitute (6), 6 and 4 by Buchwald coupling reaction to obtain 6-ethyl-3- ((3-methoxy-4- (4-(4-methylpiperazin-1-yl) piperidin-1-yl) phenylamino)-5-((tetrahydro-2H-pyran-4-yl)amino)pyrazine-2-nitrile (7), (7) is the target product gilteritinib was obtained by nitrile hydrolysis.
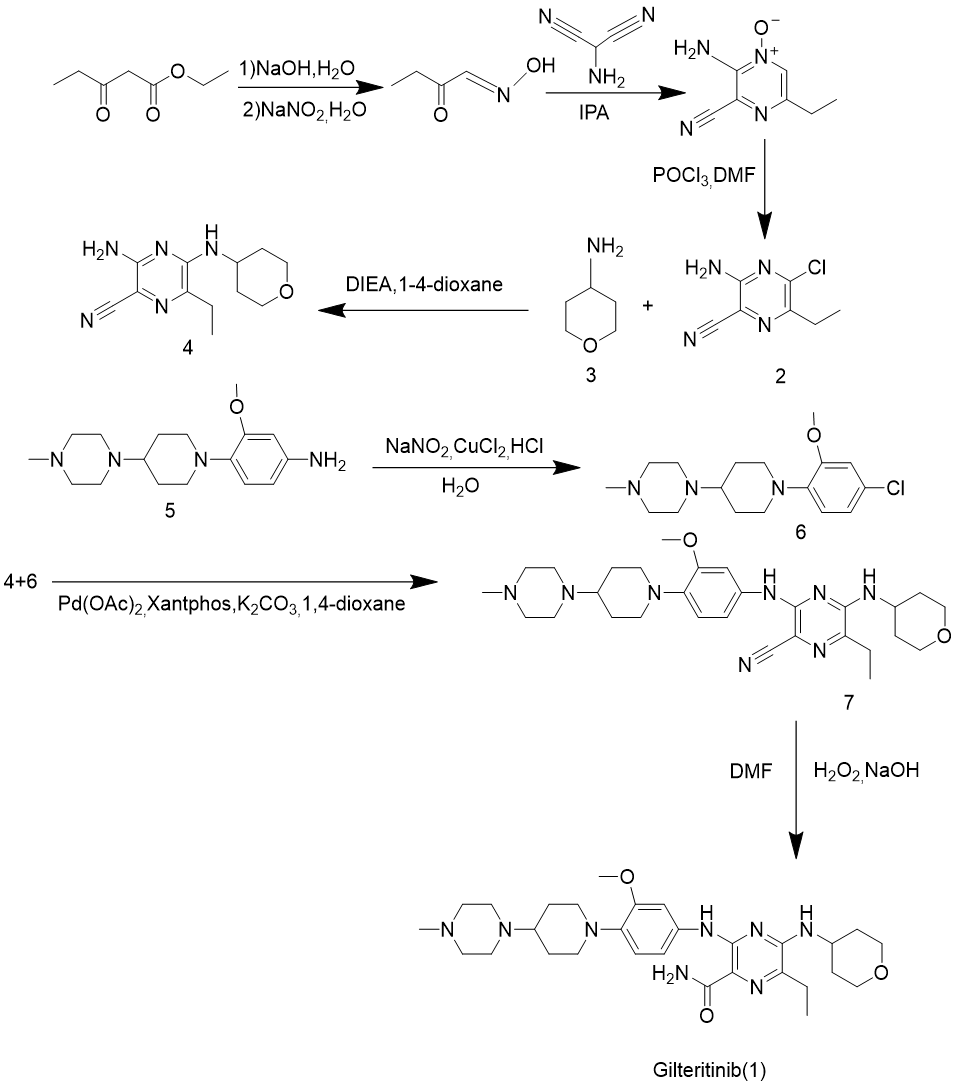
Figure 5. Gilteritinib synthesis route 3.
The reaction conditions of route 3 are relatively mild, and the generated by-products are easy to handle, and the heavy metal catalysts involved are also easy to recover. It first modifies the side chains that have not yet been connected, and then selectively replaces halogen atoms and amino groups according to the nucleophilicity of the carbon on the ring, resulting in higher efficiency. It is necessary to first use Sandmeyer to replace the amino group on a side chain raw material with a halogen atom, then use coupling reaction to connect it to the six-membered ring, and finally hydrolyze it. The convenience is average. Due to the change of the substituent on the initial six-membered ring and the change of its corresponding carbon electrophilicity, the selectivity is greatly improved. The side reaction is extremely low, which can be used in large-scale production. The atom utilization rate is 49.30%, and the purity of the product is also very high, which can reach 99.7% (HPLC method). It meets the green chemical standard and is progressiveness.
3.4. Discussion
Overall, both Route 1 and Route 2 were synthesized from six membered heterocycles with two halogen atoms, resulting in significant defects. Route 3 cleverly overcame the problem of reaction selectivity by utilizing the significant difference in electronic attraction between amino and halogen atoms. Moreover, Route 3 is excellent in other aspects, and it is not difficult to draw a conclusion. Route 3 is currently the best industrial synthesis method for gilteritinib.
Table 1. Comparison of characteristics of the three synthetic routes.
Safety | Efficiency | Convenience | Advance | |
Route 1 | poor | poor | moderate | good |
Route 2 | moderate | very poor | good | poor |
Route 3 | good | good | moderate | good |
4. Antimicrobial resistance studies of gilteritinib
4.1. Causes of drug resistance
Studies in recent years have found that some patients develop resistance after treatment with gilteritinib, which poses new challenges to treatment outcomes and patient survival. Resistance of gilteritinib is currently divided into two main types: primary drug resistance, that is, there are mutations in the resistance gene before the patient starts the drug; the other is follow-up resistance, in which patients develop resistance gene mutations after using the drug for a period of time. Primary resistance is relatively rare, accounting for about 10% of all drug resistance. Such mutations are often associated with FLT3-ITD External Alternative Struct (FEI3) mutations. These mutations reduce the drug’s affinity for the drug target, resulting in a decrease in the therapeutic effect of gilteritinib. Subsequent drug resistance is a relatively common phenomenon and one of the main problems faced by patients with AML. This usually occurs during treatment, when some leukemia cells gradually evolve a new FLT3 mutant gene, causing gilteritinib to lose some of its efficacy [1]. In addition, irregular drug use, incomplete medication, and improper drug dosage may also lead to subsequent drug resistance.
4.2. Ways to address drug resistance
However, a lot of research have been done to solve this problem, and have obtained relatively good results. The two primary approaches to addressing this issue at the moment are combination therapies and a thorough examination of the mechanism underlying gilteritinib resistance.
The mechanism of gilteritinib resistance have been identified by many researchers, Joshi et al. (2021) assessed early resistance and late resistance by using AML microenvironmental catalyzes, McMahon et al. (2019) analyzed the mutational characteristics of paired samples taken from patients to do this research [11, 12]. Although the mechanism of gilteritinib resistance has been almost thoroughly studied, only a small percentage of drugs are synthesized to replace gilteritinib, and sitravatinib is one of them [13]. More effort should be devoted to studying this method to solve the problem of drug resistance in the future.
Combination drugs have many advantages: they can play a coordinating and complementary role in targeting different pathogenesis; and can reduce each other’s adverse reactions. More importantly, it can improve patient adherence to medication. At present, there have been many literatures mentions that this method to solve the resistance of gilteritinib, and it is also one of the most mainstream solutions [11, 12, 14].
5. Conclusion
TKI is a class of complex that block tyrosine kinase's action, the authors said. It has been widely used in recent years because of its high selectivity and less side effects. It is one of the medications used to treat AML that has the FLT3 mutation. In this paper, first of all, by studying the mechanism of action of gilteritinib and receptor, this study discovered that FLT3 has two different sorts of mutations, and then analyze and compare the safety, efficiency, convenience and innovation of three synthetic pathways, and finally propose solutions to the problem of drug resistance. The issues discussed in this paper are of critical importance for the further industrial production of gilteritinib and the future treatment of AML patients, although gilteritinib has been involved in the clinical treatment of AML and a large number of researchers have done a lot of related experiments. Other somatic mutations in cancer-related genes, acquired resistance mutations in FLT3, activation of alternate signaling pathways, and other causes of resistance to FLT3 inhibitors have also been discovered, but due to the short time to market of gilteritinib, There are still some questions to be solved, and future studies should focus on gilteritinib and other drug combinations, study the therapeutic effect of gilteritinib on other leukemia types, and further explore the therapeutic use of this drug in different leukemia subtypes.
Authors Contribution
All the authors contributed equally and their names were listed in alphabetical order.
References
[1]. Perl A E, Martinelli G, Cortes J E, Neubauer A, Berman E, Paolini S, Montesinos P, Baer M R, Larson R A, Ustun C, Fabbiano F, Erba H P, Di Stasi A, Stuart R, Olin R, Kasner M, Ciceri F, Chou W-C, Podoltsev N, Recher C, Yokoyama H, Hosono N, Yoon S-S, Lee J-H, Pardee T, Fathi A T, Liu C, Hasabou N, Liu X, Bahceci E and Levis M J 2019 N. Engl. J. Med. 381 1728
[2]. Zhao J C, Agarwal S, Ahmad H, Amin K, Bewersdorf J P and Zeidan A M 2022 Blood Rev. 52 100905
[3]. Higgins A, Mangaonkar A A, Hefazi M, Viswanatha D, Horna P, Foran J and Gangat N 2018 Leuk. Res. 72 1
[4]. Li K X, Pan W Y, Wu S J and Huang Y X 2021 J. Clin. Hematol. 34 202
[5]. Wu M, Li C and Zhu X 2018 J. Hematol. Oncol. 11 133
[6]. Mori M, Kaneko N, Ueno Y, Yamada M, Tanaka R, Saito R, Shimada I, Mori K and Kuromitsu S 2017 Invest. New Drugs 35 556
[7]. Kawase T, Nakazawa T, Eguchi T, Tsuzuki H, Ueno Y, Amano Y, Suzuki T, Mori M and Yoshida T 2019 Oncotarget 10 6111
[8]. Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, Lippke J and Saxena K 2004 Mol. Cell 13 169
[9]. Ayala-Aguilera C C, Valero T, Lorente-Macías Á, Baillache D J, Croke S and Unciti-Broceta A 2022 J. Med. Chem. 65 1047
[10]. Liu K, Li Z N, Zhong Y and Liu Y 2022 Chinese J. Med. Chem. 32 915
[11]. Joshi S K, Nechiporuk T, Bottomly D, Piehowski P D, Reisz J A, Pittsenbarger J, Kaempf A, Gosline S J C, Wang Y-T, Hansen J R, Gritsenko M A, Hutchinson C, Weitz K K, Moon J, Cendali F, Fillmore T L, Tsai C-F, Schepmoes A A, Shi T, Arshad O A, McDermott J E, Babur O, Watanabe-Smith K, Demir E, D’Alessandro A, Liu T, Tognon C E, Tyner J W, McWeeney S K, Rodland K D, Druker B J and Traer E 2021 Cancer Cell 39 999
[12]. McMahon C M, Ferng T, Canaani J, Wang E S, Morrissette J J D, Eastburn D J, Pellegrino M, Durruthy-Durruthy R, Watt C D, Asthana S, Lasater E A, DeFilippis R, Peretz C A C, McGary L H F, Deihimi S, Logan A C, Luger S M, Shah N P, Carroll M, Smith C C and Perl A E 2019 Cancer Discov. 9 1050
[13]. Zhang Y, Wang P, Wang Y and Shen Y 2023 Biomark Res. 11 8
[14]. Daver N, Perl A E, Maly J, Levis M, Ritchie E, Litzow M, McCloskey J, Smith C C, Schiller G, Bradley T, Tiu R V, Naqvi K, Dail M, Brackman D, Siddani S, Wang J, Chyla B, Lee P and Altman J K 2022 J. Clin. Oncol. 40 4048
Cite this article
Huang,R.J.;Jiang,Z.Y.;Lin,K. (2023). A study of the synthetic route and resistance of gilteritinib. Theoretical and Natural Science,15,255-262.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Perl A E, Martinelli G, Cortes J E, Neubauer A, Berman E, Paolini S, Montesinos P, Baer M R, Larson R A, Ustun C, Fabbiano F, Erba H P, Di Stasi A, Stuart R, Olin R, Kasner M, Ciceri F, Chou W-C, Podoltsev N, Recher C, Yokoyama H, Hosono N, Yoon S-S, Lee J-H, Pardee T, Fathi A T, Liu C, Hasabou N, Liu X, Bahceci E and Levis M J 2019 N. Engl. J. Med. 381 1728
[2]. Zhao J C, Agarwal S, Ahmad H, Amin K, Bewersdorf J P and Zeidan A M 2022 Blood Rev. 52 100905
[3]. Higgins A, Mangaonkar A A, Hefazi M, Viswanatha D, Horna P, Foran J and Gangat N 2018 Leuk. Res. 72 1
[4]. Li K X, Pan W Y, Wu S J and Huang Y X 2021 J. Clin. Hematol. 34 202
[5]. Wu M, Li C and Zhu X 2018 J. Hematol. Oncol. 11 133
[6]. Mori M, Kaneko N, Ueno Y, Yamada M, Tanaka R, Saito R, Shimada I, Mori K and Kuromitsu S 2017 Invest. New Drugs 35 556
[7]. Kawase T, Nakazawa T, Eguchi T, Tsuzuki H, Ueno Y, Amano Y, Suzuki T, Mori M and Yoshida T 2019 Oncotarget 10 6111
[8]. Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, Lippke J and Saxena K 2004 Mol. Cell 13 169
[9]. Ayala-Aguilera C C, Valero T, Lorente-Macías Á, Baillache D J, Croke S and Unciti-Broceta A 2022 J. Med. Chem. 65 1047
[10]. Liu K, Li Z N, Zhong Y and Liu Y 2022 Chinese J. Med. Chem. 32 915
[11]. Joshi S K, Nechiporuk T, Bottomly D, Piehowski P D, Reisz J A, Pittsenbarger J, Kaempf A, Gosline S J C, Wang Y-T, Hansen J R, Gritsenko M A, Hutchinson C, Weitz K K, Moon J, Cendali F, Fillmore T L, Tsai C-F, Schepmoes A A, Shi T, Arshad O A, McDermott J E, Babur O, Watanabe-Smith K, Demir E, D’Alessandro A, Liu T, Tognon C E, Tyner J W, McWeeney S K, Rodland K D, Druker B J and Traer E 2021 Cancer Cell 39 999
[12]. McMahon C M, Ferng T, Canaani J, Wang E S, Morrissette J J D, Eastburn D J, Pellegrino M, Durruthy-Durruthy R, Watt C D, Asthana S, Lasater E A, DeFilippis R, Peretz C A C, McGary L H F, Deihimi S, Logan A C, Luger S M, Shah N P, Carroll M, Smith C C and Perl A E 2019 Cancer Discov. 9 1050
[13]. Zhang Y, Wang P, Wang Y and Shen Y 2023 Biomark Res. 11 8
[14]. Daver N, Perl A E, Maly J, Levis M, Ritchie E, Litzow M, McCloskey J, Smith C C, Schiller G, Bradley T, Tiu R V, Naqvi K, Dail M, Brackman D, Siddani S, Wang J, Chyla B, Lee P and Altman J K 2022 J. Clin. Oncol. 40 4048