







1. Introduction
According to 2022 statistics and projections for new cancer cases and deaths by the American Cancer Society, lung cancer has become the leading cause of malignant tumor deaths in the United States [1]. Lung cancer can be categorized into Small Cell Lung Cancer and Non-Small Cell Lung Cancer (NSCLC), accounting for 15% and 85% of lung cancer cases, respectively [2]. NSCLC can further be subdivided into adenocarcinoma, squamous cell carcinoma, and large cell carcinoma [3]. In the progression of NSCLC, the Epidermal Growth Factor Receptor (EGFR) plays a critical role. Overexpression of EGFR has been found in all three subtypes of NSCLC, with a prevalence rate of up to 90% in squamous cell carcinoma [4]. As a result, drugs targeting EGFR have become a hopeful avenue for treating NSCLC. Gefitinib, a representative of the first-generation epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs), can compete for ATP binding sites in the EGFR-TKI catalytic domain, blocking cellular signal transduction and thereby inhibiting the proliferation and differentiation of cancer cells [5]. The mode of action, target sites, regulatory mechanisms, and involved signaling pathways of gefitinib are currently the focus of research.
2. Incidence and Mechanism of Lung Cancer
The incidence of lung cancer is associated with various factors. Surveys indicate that the incidence rates of lung cancer differ among various ethnic groups. For example, in the United States, Compared to other racial groupings, Asian Americans had lower lung cancer incidence and fatality rates [6]. Smoking has been identified as a significant factor affecting lung cancer incidence. The incidence and mortality rates of lung cancer are much higher in developed countries compared to developing countries. A few decades ago, smoking rates in these countries peaked; however, current incidence and mortality rates have begun to decline. In contrast, in tobacco-prevalent developing countries, both figures are on the rise [7].
Extensive research has shed light on the mechanisms underlying lung cancer. A close relationship exists between EGFR and the onset of lung cancer. In its inactive or dormant state, EGFR exists as a monomer on the cell membrane, and the Receptor Tyrosine Kinase (RTK) also exists as a monomer in a non-active state. Once a signaling molecule binds to the extracellular domain of the receptor, dimerization occurs on the membrane, leading to contact between the intracellular domains' tails. This activates the protein kinase and results in its phosphorylation, forming a signaling complex. A dimerization of EGFR monomers with other EGFR molecules or other ErbB family protein receptors occurs when the extracellular region of EGFR binds with extracellular ligands. The tyrosine kinase region of the EGFR cell binds to one molecule of ATP after dimerization, and upon phosphorylation, this region is active. Specific tyrosine residues on the C-terminal then experience phosphorylation through transphosphorylation or autophosphorylation. These phosphorylation sites operate as binding sites for additional downstream signaling molecules and as signaling pathway activators, facilitating the proliferation, differentiation, and migration of tumor cells [5].
3. Action Sites of Gefitinib
In the traditional drug discovery process, chemists supply compounds, while biologists utilize tissues, cells, or animals for screening. Through this approach, numerous classic drugs, such as penicillin, have been discovered. In contrast, modern drug discovery has evolved to include biochemists using purified proteins (targets) for affinity screening of compounds. Furthermore, the focus of research has shifted from the relationship between "drug and disease" to that of "drug and target." In summary, the key to modern drug discovery lies in targeting specific proteins.
Gefitinib is an EGFR-TKIs drug that targets the ATP cleft within the epidermal growth factor receptor, preventing the receptor's autophosphorylation. This action inhibits downstream signaling pathways, leading to cell stasis or cell death, ultimately achieving a therapeutic effect on lung cancer [8].
According to case records, Gefitinib shows therapeutic effects for certain autoimmune diseases such as rheumatoid arthritis [9]. A 72-year-old female patient with NSCLC and osteoarthritis underwent treatment with Gefitinib. Two weeks later, her joint pain and stiffness had significantly improved. However, when the drug was temporarily discontinued due to diarrhea, the symptoms reappeared [10]. Experiments suggest that the off-target effects of Gefitinib, primarily used for cancer treatment, might endow it with the capability to treat autoimmune and other non-autoimmune inflammatory diseases [9]. Due to the lengthy approval process and high development costs of new drugs, repurposing existing medications for the treatment of other diseases is a viable approach. Further experimental and clinical studies are warranted to establish Gefitinib as a potential drug for psoriasis and rheumatoid arthritis.
4. Synthesis Route of Gefitinib
To facilitate a better understanding of the variations in the synthesis routes of Gefitinib, the following will present these routes in chronological order.
The first synthesis route was proposed by Gibson in 1996, as shown in Figure 1. Figure 1 depicts the Gibson-reported synthesis method. The expensive starting material 6,7-dimethoxyquinazolin-4(3H)-one (1) is first subjected to region-selective demethylation, which is followed by O-protection. Sulfonyl chloride or phosphoryl chloride are then used to prepare the necessary chlorinated chemicals. The final molecule, Gefitinib, is produced through further coupling with 3-chloro-4-fluoroaniline and the addition of the remaining functional groups required for compound 7 [11].
In 2009, Gilday reported a Gefitinib synthesis route that starts with 3-hydroxy-4-methoxybenzaldehyde as the initial material, as illustrated in Figure 2. The 3-hydroxy-4-methoxybenzaldehyde is first converted into the corresponding nitrile, followed by alkylation, nitration, reduction, nitrile hydrolysis, cyclization, and chlorination. The chloride is then treated with 3-chloro-4-fluoroaniline. Through these 8 processes, Gefitinib is synthesized [12].
A synthesis route for Gefitinib starting from 3-hydroxy-4-methoxybenzoic acid methyl ester was reported by P Gong in 2005, as shown in Figure 3. Gefitinib is synthesized through a seven-step reaction route [13].
All three of the aforementioned synthetic routes are traditional chemical methods. In actual production, numerous challenges are encountered. These methods have been observed to be prone to production safety incidents, possess complex purification steps, and involve lengthy synthetic routes. Additionally, the reagents used in production could potentially cause environmental damage. For example, in the production of paclitaxel using Taxus as raw material, the entire process requires 34 steps, resulting in a final yield of only 0.44%. Such methods do not conform to the principles of green chemistry, which advocates for maximizing atomic economy.
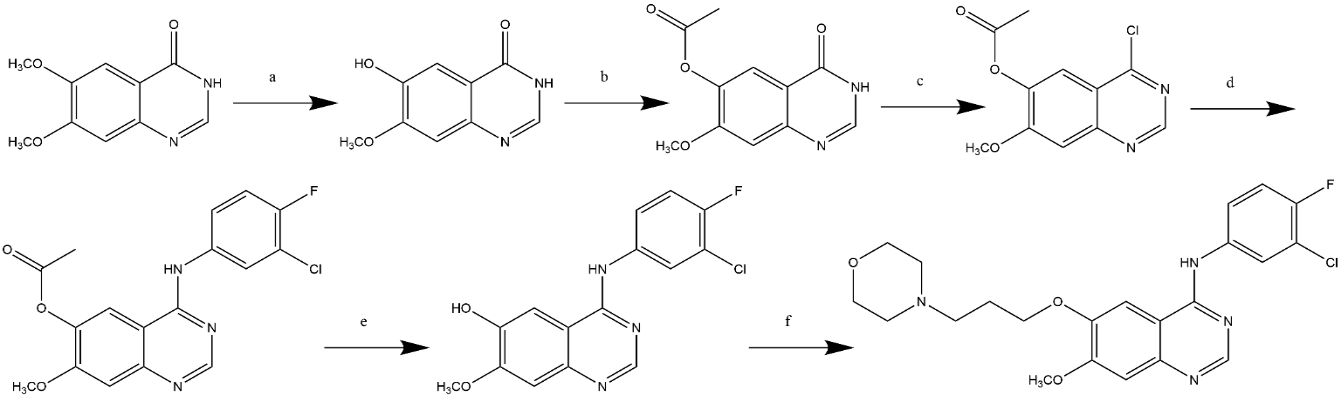
Figure 1. Gibson's Synthetic Route.
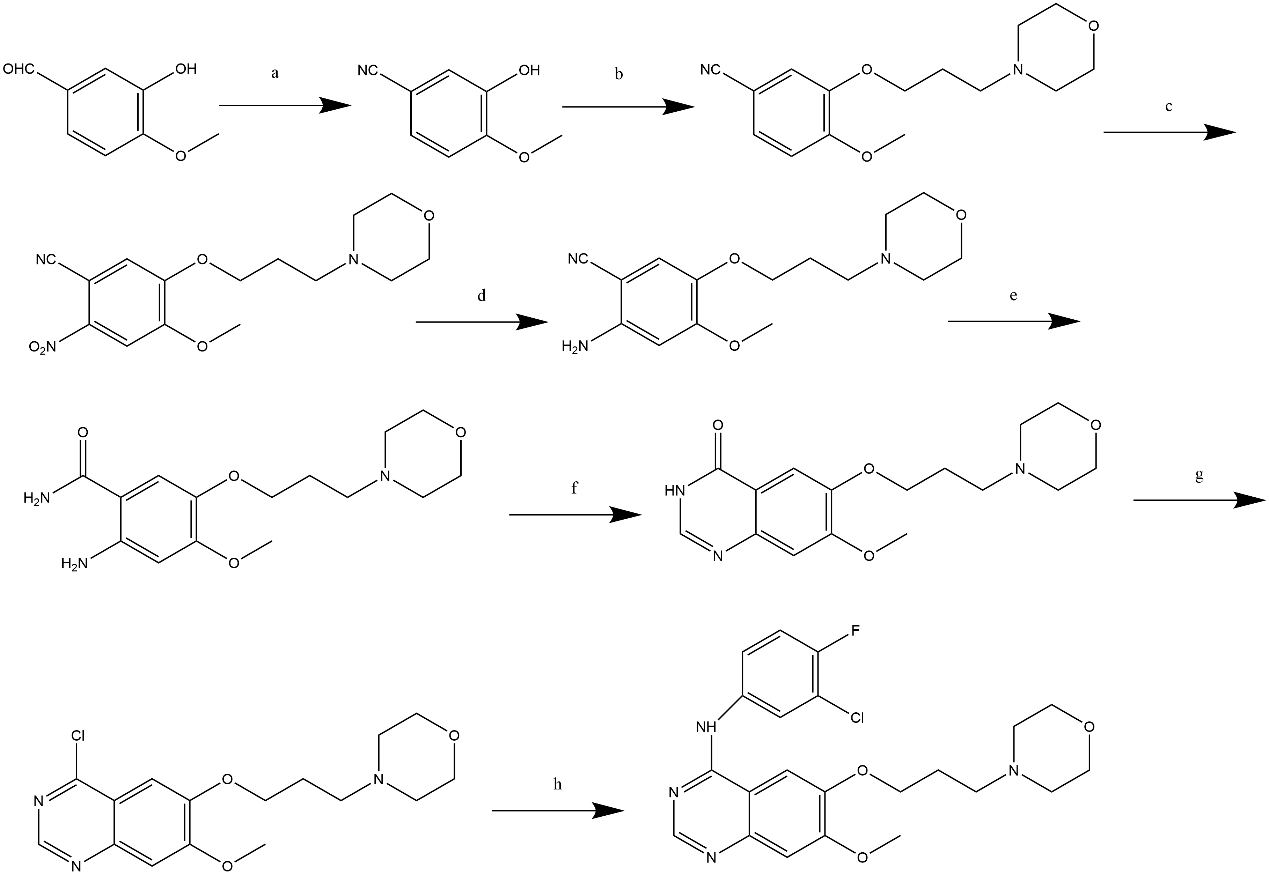
Figure 2. Gilday's Synthetic Route.
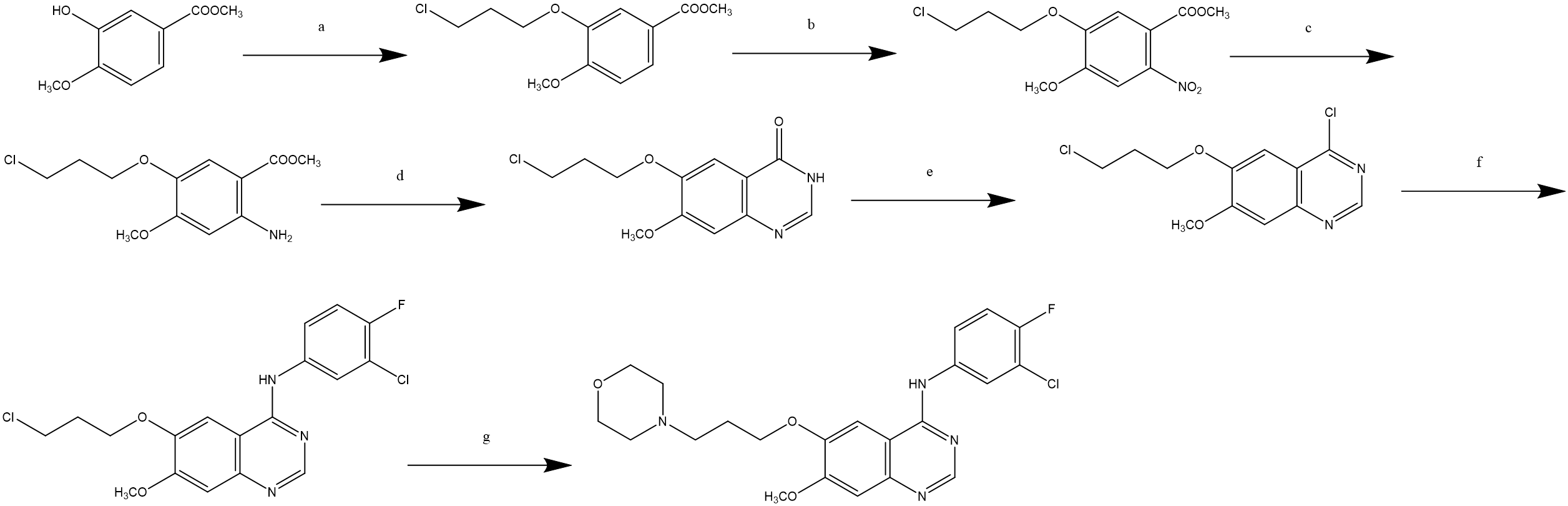
Figure 3. P Gong's Synthetic Route.
5. Prospects for Future Synthesis Routes
Synthetic methods for other drugs have provided insights for the synthesis of Gefitinib. In designing synthetic routes for organic pharmaceuticals, the main task identified by the researchers is to imaginatively seek feasible approaches among a myriad of reactions. The goal is to compile these into conceptual principles and methods that guide the synthetic design, thereby devising an appropriate synthetic route.
Five major strategies commonly used in synthetic route design have been summarized: transform-based strategies, structural-goal strategies, topological strategies, stereochemical strategies and functional group-based strategies. Additionally, several points need to be analyzed. First, the identification and confirmation of functional groups present in the target molecule are required. Second, consideration must be given to where to make cuts, and which known and reliable reactions can be utilized for this purpose. Lastly, an analysis of the fragments should be conducted to see if readily available starting materials can be identified. Through these methods, significant time can be saved, and rational synthetic routes can be designed.
Due to the inevitable challenges that arise with each generation of targeted drugs during treatment, there is a continuous need for innovation to design new medications that address the limitations of their predecessors. The researchers have identified several methods suitable for the discovery of new drugs: computer-aided drug design (CADD), DNA encoded compound library, fragment-based drug design and structure-based drug design.
Rational drug design is crucial for the generation of new pharmaceuticals. As such, Computer-Aided Drug Design is an indispensable component in current new drug development efforts. CADD enables the application of effective and efficient strategies to overcome barriers in the drug design domain, thereby aiding in the accurate design and development of new drugs. This, in turn, saves substantial time and resources.
In recent years, with the continuous integration of chemical and biological synthesis, enzyme-catalyzed synthesis methods have attracted significant attention. The combination of chemical and biological synthesis can achieve ideal synthesis results. For example, in the production of Sitagliptin and MK-8591, the new enzyme-catalyzed route for Sitagliptin compared to the traditional chemical route has fewer steps, higher yields, an increase in production capacity of 53%, and a 19% reduction in waste [14]. In comparison to chemical synthesis, which requires 16 steps and nine separations with a final yield of 14%, the enzyme-catalyzed synthesis for MK-8591 needs only five steps and two separations, resulting in a high final yield of 51% [15]. These enzyme-catalyzed routes not only enable production at ambient temperature and pressure, fulfilling the requirement for higher energy efficiency, but also minimize waste generation, perfectly aligning with the principles of green chemistry.
With advancements in technology, bioelectrocatalysis, which incorporates biocatalysis, has successfully solved the problem of sustainable drug synthesis [16]. This also provides new perspectives for the synthesis of Gefitinib. Therefore, it is hoped that future production of Gefitinib will employ new technological approaches like enzyme catalysis and bioelectrochemical synthesis. This would not only increase its production but also promote environmentally friendly drug production methods, thus resolving the environmental, economic, and safety issues associated with traditional synthesis methods.
6. Current Applications and Problems of Gefitinib
Gefitinib, a first-generation EGFR TKI, has been approved by the FDA and EMA for use in lung cancer caused by EGFR mutations. However, nearly all patients experience adverse reactions to Gefitinib, such as developing resistance to targeted therapy around the 10th to 12th month [8]. This diminishes the therapeutic effects. Several acquired resistance mechanisms have been identified, but the most common is the gatekeeper mutation T790M found in the pocket where EGFR binds to ATP. This mutation confers resistance to Gefitinib in 50-60% of patients [17]. The T790M mutation reduces the affinity of EGFR TKIs for EGFR and increases the affinity of ATP for EGFR, thus enhancing the survival capability of NSCLC cells with the T790M mutation [18]. Currently, there are four methods to overcome the resistance to Gefitinib. The first is the use of third-generation EGFR TKIs like Osimertinib, although new resistance often develops after 9-13 months of treatment [19]. The second method involves deactivating bypass signals using small molecule inhibitors or monoclonal antibodies, but its clinical application is restricted by the complexity of the production process [20]. The third method employs CuS nanoparticles as photodynamic nano-switches that specifically deactivate overly active bypass signaling in resistant tumor cells without interfering with the same signaling pathways in normal cells. In experiments, localized elevation of reactive oxygen species (ROS) levels in tumor cells under infrared laser irradiation led to blockade of the IGF1R and its downstream AKT/ERK/NF-κB signaling cascade, making the tumor cells more sensitive to Gefitinib and prolonging the lifespan of experimental mice [20]. The fourth method involves structural modifications at key positions of the quinazoline core [21]. For example, new EGFR TKIs created by adding cinnamoyl amide to the quinazoline scaffold can be used to reverse the resistance caused by the T790M mutation. Structure-activity relationship (SAR) studies show that methoxy and acyloxy substitutions on the phenyl ring of cinnamic acid can improve activity, with the highest inhibition against T790M being up to 11 times more effective than Gefitinib [22].
Adverse effects brought about by first-generation EGFR TKIs are generally mild, yet when they occur, they can impact the quality of life for patients. Rash and diarrhea are two of these medications' most frequent Grade 3 or Grade 4 side effects, but less than 10% of patients stop taking them as a result of side effects [23].
In terms of pharmacodynamics, subjects were divided into two groups: healthy volunteers and NSCLC patients. Both groups were administered 250mg of Gefitinib orally. The bioavailability of Gefitinib was found to be 57% in the volunteer group and 59% in the patient group. Five hours after oral administration of Gefitinib, the mean peak plasma concentration in the volunteer group was 85 nM/mL, while in the patient group it was 159 nM/mL. Experimental data indicated that bioavailability was independent of dose, and food had no significant clinical impact on the utilization of Gefitinib. According to the blood concentration curve after oral administration, it was observed that Gefitinib has a rapid plasma clearance rate and a wide distribution range [24]. Therefore, despite Gefitinib's absolute bioavailability reaching approximately 50%, its pharmacodynamic characteristics still support a once-daily dosing regimen [25].
Although first-generation EGFR TKIs such as Gefitinib have shown improvements in progression-free survival when compared to chemotherapy drugs represented by platinum compounds, an extension in overall survival has not been achieved [26]. Therefore, immunotherapy needs to be incorporated into subsequent treatments to prolong overall survival [27]. Another concern, as mentioned earlier, is the off-target effects that Gefitinib might possess, thereby limiting its therapeutic efficacy [25].
Due to the hydrophobic nature of Gefitinib, its accumulation levels in the skin, kidneys, and liver are considerably higher than in the plasma, which could lead to its deposition in normal tissues like bone marrow [28].
Combining Gefitinib with certain drugs for treating NSCLC can lead to severe consequences. For example, in an experiment where Gefitinib was combined with Vincristine, symptoms of neutropenia were observed potentially endangering patients' lives [29].
7. Conclusion
EGFR TKIs are currently the standard first-line therapy for late-stage NSCLC patients. This article has provided an overview of the sites of action, involved signaling pathways, and regulatory mechanisms of Gefitinib. It also explained from a genetic perspective how resistance to Gefitinib occurs, and listed several methods to overcome this resistance. It is important to note that no method has been found yet to completely eliminate tumor cell resistance to Gefitinib, so this is considered the next key focus for research on Gefitinib. Additionally, the adverse effects produced by Gefitinib during treatment could potentially have a detrimental impact on patients, and it is hoped that modifications to the drug itself could reduce its residual levels in the body.
The researchers assert that along with focusing on treatment modalities, prevention strategies should also be emphasized. Firstly, current treatments for lung cancer primarily involve chemotherapy and radiation, which have low survival rates and adverse effects on the patient's body. Targeted therapy has shown promise for curing lung cancer, yet issues such as acquired resistance and off-target effects still exist. Secondly, the growing number of lung cancer patients leads to the consumption of more medical resources, which impacts the treatment of other patients. In contrast, prevention measures for lung cancer are considerably simpler. Avoiding contact and inhalation of carcinogens, such as dioxins, can be effective. Correcting unhealthy lifestyle habits can also significantly reduce the risk of developing lung cancer. Additionally, it should be noted that the prevalence of electronic cigarettes has also led to an increased incidence of lung cancer among younger populations.
References
[1]. Siegel R L, Miller K D, Fuchs H E and et al. 2022 CA. Cancer J. Clin. 72(1) 7–33
[2]. Remark R, Becker C, Gomez J E, and et al. 2015 Am. J. Respir. Crit. Care Med. 191(4) 377-390
[3]. Pikor L A, Ramnarine V R, Lam W L, and et al. 2013 Lung Cancer Amst. Neth. 82(2) 179–189
[4]. Mansour M A, Abbas S, Abdel-Rahman H M, and et al. 2023 RSC Adv. 13(27) 18825–18853
[5]. Peng W, Pan Q, Ye J, Fang Y, and et al. 2023 Front. Oncol. 13 1120278
[6]. Lei F, Zheng Y and Li C 2022 J. Immigr. Minor. Health 24(2) 526–545
[7]. Leiter A, Veluswamy R R and Wisnivesky J P 2023 Nat. Rev. Clin. Oncol. 20(9) 624–639
[8]. Solassol I, Pinguet F and Quantin X 2019 Biomolecules 9(11) 668
[9]. Brooks M B 2013 The Oncologist 18(1) e3–e5
[10]. Moryl N, Obbens E A, Ozigbo O H, and Kris M G 2006 J. Support. Oncol. 4(3) 111
[11]. Gibson K H 1996 Quinazoline Derivatives (Patent WO9633980)
[12]. Peter G J and David M 2009 Process for the Preparation of 4- (3’chloro-4’-Fluoroanilino) -7-Methoxy-6- (3-Morpholinopropoxy) Quinazoline (Patent CN101348471)
[13]. Li Y, Yong Z, Ping G and et al. 2005 Chin. J. Med. Chem. 1 3
[14]. Savile C K, Colbeck J C, Huisman G W, and et al. 2010 Science 329(5989) 305–309
[15]. Huffman M A, Fryszkowska A, Campos K R, and et al. 2019 Science 366(6470) 1255–1259
[16]. Boucher D G, Nguyen Z A, Minteer S D, and et al. 2023 Angew. Chem. Int. Ed Engl. 2023 e202307780
[17]. Wu S and Shih J 2018 Mol. Cancer 17(1) 38
[18]. Saldaña-Rivera L, Bello M and Méndez-Luna D 2019 J. Biomol. Struct. Dyn. 37(17) 4671–4684
[19]. Wang S, Song Y and Liu D 2016 Front. Med. 10(4) 383–388
[20]. Gu X, Lin M, Fan C, and et al. 2019 Nano Lett. 19(5) 3344–3352
[21]. Șandor A, Oniga I, Oniga O, and et al. 2023 Pharmaceuticals 16(4) 534
[22]. Zhang, B Xu Z, Liu Q, and et al. 2021 Bioorganic Chem. 117 105420
[23]. Ding P N, Lord S J, Gebski, V and et al. 2017 J. Thorac. Oncol. 12(4) 633–643
[24]. Swaisland H C, Laight A, Ranson M, and et al. 2005 Clin. Pharmacokinet. 44(11) 1165–1177
[25]. Zhao C, Han S and Li P 2017 Curr. Drug Deliv. 14(2) 282–288
[26]. Brückl W, Tufman A and Huber R M 2017 Expert Rev. Anticancer Ther. 17(2) 143–155
[27]. Araghi M, Heidarnejad maleki A, Rostami S, and et al. 2023 Cancer Cell Int. 23 162
[28]. Cui J, Guo W, Wang C, and et al. 2007 J. Control Release Soc. 118(2) 204–215
[29]. Yoshimura M, Imamura F, Yamamoto S, and et al. 2004 Lung Cancer Amst. Neth. 45(1) 121–123
Cite this article
Chen,F. (2023). Evaluation of gefitinib based on indicators of targeted drugs. Theoretical and Natural Science,16,48-54.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Siegel R L, Miller K D, Fuchs H E and et al. 2022 CA. Cancer J. Clin. 72(1) 7–33
[2]. Remark R, Becker C, Gomez J E, and et al. 2015 Am. J. Respir. Crit. Care Med. 191(4) 377-390
[3]. Pikor L A, Ramnarine V R, Lam W L, and et al. 2013 Lung Cancer Amst. Neth. 82(2) 179–189
[4]. Mansour M A, Abbas S, Abdel-Rahman H M, and et al. 2023 RSC Adv. 13(27) 18825–18853
[5]. Peng W, Pan Q, Ye J, Fang Y, and et al. 2023 Front. Oncol. 13 1120278
[6]. Lei F, Zheng Y and Li C 2022 J. Immigr. Minor. Health 24(2) 526–545
[7]. Leiter A, Veluswamy R R and Wisnivesky J P 2023 Nat. Rev. Clin. Oncol. 20(9) 624–639
[8]. Solassol I, Pinguet F and Quantin X 2019 Biomolecules 9(11) 668
[9]. Brooks M B 2013 The Oncologist 18(1) e3–e5
[10]. Moryl N, Obbens E A, Ozigbo O H, and Kris M G 2006 J. Support. Oncol. 4(3) 111
[11]. Gibson K H 1996 Quinazoline Derivatives (Patent WO9633980)
[12]. Peter G J and David M 2009 Process for the Preparation of 4- (3’chloro-4’-Fluoroanilino) -7-Methoxy-6- (3-Morpholinopropoxy) Quinazoline (Patent CN101348471)
[13]. Li Y, Yong Z, Ping G and et al. 2005 Chin. J. Med. Chem. 1 3
[14]. Savile C K, Colbeck J C, Huisman G W, and et al. 2010 Science 329(5989) 305–309
[15]. Huffman M A, Fryszkowska A, Campos K R, and et al. 2019 Science 366(6470) 1255–1259
[16]. Boucher D G, Nguyen Z A, Minteer S D, and et al. 2023 Angew. Chem. Int. Ed Engl. 2023 e202307780
[17]. Wu S and Shih J 2018 Mol. Cancer 17(1) 38
[18]. Saldaña-Rivera L, Bello M and Méndez-Luna D 2019 J. Biomol. Struct. Dyn. 37(17) 4671–4684
[19]. Wang S, Song Y and Liu D 2016 Front. Med. 10(4) 383–388
[20]. Gu X, Lin M, Fan C, and et al. 2019 Nano Lett. 19(5) 3344–3352
[21]. Șandor A, Oniga I, Oniga O, and et al. 2023 Pharmaceuticals 16(4) 534
[22]. Zhang, B Xu Z, Liu Q, and et al. 2021 Bioorganic Chem. 117 105420
[23]. Ding P N, Lord S J, Gebski, V and et al. 2017 J. Thorac. Oncol. 12(4) 633–643
[24]. Swaisland H C, Laight A, Ranson M, and et al. 2005 Clin. Pharmacokinet. 44(11) 1165–1177
[25]. Zhao C, Han S and Li P 2017 Curr. Drug Deliv. 14(2) 282–288
[26]. Brückl W, Tufman A and Huber R M 2017 Expert Rev. Anticancer Ther. 17(2) 143–155
[27]. Araghi M, Heidarnejad maleki A, Rostami S, and et al. 2023 Cancer Cell Int. 23 162
[28]. Cui J, Guo W, Wang C, and et al. 2007 J. Control Release Soc. 118(2) 204–215
[29]. Yoshimura M, Imamura F, Yamamoto S, and et al. 2004 Lung Cancer Amst. Neth. 45(1) 121–123