







1. Introduction
Ferroptosis is a type of regulated cell death, distinct from apoptosis, that is initiated through iron-mediated phospholipid peroxidation [1]. Ferroptosis is a form of regulated cell death characterized by its dependence on iron and increased lipid peroxidation. It is triggered when levels of the enzyme GPX4, which protects against phospholipid hydroperoxidation, are depleted, when redox-active iron accumulates to excess, and when phospholipids containing polyunsaturated fatty acids (PUFAs) undergo oxidative modification. The interplay between these factors - iron accumulation, loss of GPX4 activity, and peroxidation of PUFA-containing phospholipids - induces ferroptosis [2]. The cellular processes that regulate ferroptosis integrate a variety of signals, encompassing both pro-survival and pro-death cues from subcellular organelles, to ultimately decide whether to activate this lethal pathway or not [3]. Targeted cell death is a prevalent strategy in cancer treatment. Given that ferroptosis inducers have the potential to specifically target cancer cells, incorporating ferroptosis-inducing medications can enhance the anti-tumor effectiveness of these drugs [4]. In current cancer treatment, clinical compounds induce ferroptosis mainly by suppressing System Xc- and GPX4. Additionally, some natural products can stimulate ferroptosis specifically in cancer cells, underscoring their importance. The combined effects of synthetic inhibitors and natural inducers that target these two key proteins represent the major mechanisms for exploiting ferroptosis therapeutically.
In this review, this summarizes our current comprehension of the regulatory factors that govern ferroptosis, the interactions involving ferroptosis and particular organelles, as well as the connections between apoptosis and ferroptosis. In conclusion, several drug examples are associated with these mechanisms. It is anticipated that this review will contribute to an improved comprehension of ferroptosis and provide the groundwork for future investigations and the advancement of cancer therapies based on ferroptosis.
2. Ferroptosis
Ferroptosis is a specific type of regulated cell death that is distinct from apoptosis. It constitutes a novel form of cell demise with characteristic features that differentiate it from other modes of programmed cell death. The properties that define ferroptosis make it a unique cell death pathway separate from apoptotic and other cell death programs. It is primarily marked by its dependence on iron-induced phospholipid peroxidation [1]. Ferroptosis can be characterized by two main pathways: the extrinsic route, relying on transporters, and the intrinsic route, governed by enzymes [5]. Ferroptosis can take place in both cancer cells and normal cells; however, selectively inducing ferroptosis in tumors while safeguarding normal tissues remains a challenge. Ferroptosis assumes a pivotal role in regulating the initiation and evolution of various diseases, making it a focal point of research and a subject of keen interest for improving the management and prognosis of these related conditions (Figure 1).
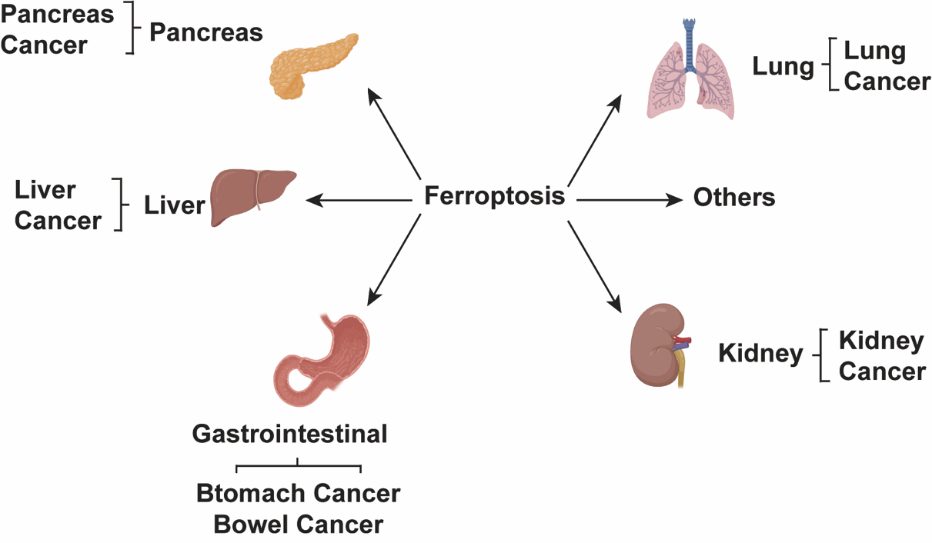
Figure 1. Ferroptosis has assumed significant roles in the pathogenesis of various systemic diseases.
2.1. The characteristics of ferroptotic cell death
Ferroptosis is a type of regulated cell death that relies on iron accumulation and elevated lipid peroxidation. The key features that define ferroptosis are its requirement for iron and accumulation of oxidized lipid damage. Ferroptosis is characterized by an impaired capacity to repair lipid peroxides. The dependency of ferroptosis on iron and lipid peroxidation arises from depleted levels of the lipid repair enzyme GPX4, accumulation of redox-active iron, and iron-mediated oxidation of phospholipids containing polyunsaturated fatty acids. The combination of deficient GPX4 activity, iron excess, and resulting phospholipid damage underlies the reduced lipid repair during ferroptosis [2]. Cells undergoing ferroptosis exhibit morphological characteristics that are unique compared to other modes of cell demise. Hallmarks of ferroptosis include smaller mitochondria with compacted membranes and disappearance of mitochondrial cristae. These distinct mitochondrial changes give ferroptotic cells a characteristic ultrastructural appearance that sets them apart. These mitochondrial changes give ferroptotic cells a distinctive appearance [6].
2.2. Methods to promote and prevent ferroptosis
Surplus iron is sequestered within ferritin and can be transported to lysosomes for degradation via the function of NCOA4, leading to the liberation of iron. This process, referred to as ferritinophagy, can promote the initiation of ferroptosis [7]. The onset of ferroptosis can be averted by employing iron chelators, while introducing exogenous iron intensifies ferroptosis [8].
3. The relationship between ferroptosis and specific cell organelles
The cellular apparatus orchestrating ferroptosis assimilates diverse signals from subcellular organelles, encompassing both pro-survival and pro-death cues, before determining whether to initiate the lethal pathway or not [3].
3.1. Ferroptosis with endoplasmic reticulum
The endoplasmic reticulum (ER) exhibits a high degree of sensitivity to alterations in the intracellular milieu, including changes in pH and viscosity. Even slight variations can elicit stress responses within the ER [9]. Ferroptosis is an exceptional, non-apoptotic pathway of cell demise linked to diseases connected with endoplasmic reticulum stress [10]. Viscosity, as a pivotal physicochemical factor, is intricately connected to diffusion-driven mechanisms, exerts significant influence on the control of various biological processes, and serves as a vital gauge for discerning numerous subcellular biological activities [11]. During the ferroptosis process, an iron-dependent Fenton reaction takes place, generating lipid radicals. Subsequently, unsaturated lipids undergo conversion into lipid peroxides through radical-initiated reactions.
3.2. Ferroptosis with mitochondria
Mitochondria assume a crucial role in the context of ferroptosis initiated by cysteine deficiency. However, mitochondrial abnormalities are not critical for ferroptosis induced by inhibition of glutathione peroxidase-4 (GPX4), the terminal effector in the ferroptotic pathway. Iron can be transported into mitochondria by SLC25A37 and SLC25A28 for heme/Fe-S cluster biosynthesis or stored in mitochondrial ferritin. But mitochondrial dysfunction is not an essential feature of GPX4 inhibition-induced ferroptosis. Heme, when catalyzed by HMOX1, decomposes into Fe2+ and plays a critical role as a cofactor in the electron transport chain (ETC), thereby supporting ferroptosis. The suppression of proteins engaged in iron-sulfur cluster formation, including NFS1, FXN, and ISCU, can intensify ferroptosis by provoking an iron-deprivation response mediated by IREB2. On the other hand, iron-sulfur (Fe-S) proteins CISD1 and CISD2 mitigate ferroptosis by lowering intracellular iron levels [3]. Mechanistically, cysteine deficiency leads to hyperpolarization of the mitochondrial membrane potential and accumulation of lipid peroxides. Inhibition of either the mitochondrial tricarboxylic acid (TCA) cycle or electron transport chain (ETC) can normalize the hyperpolarized mitochondrial membrane potential, decrease lipid peroxide buildup, and suppress ferroptosis. The mitochondrial hyperpolarization and lipid peroxidation induced by cysteine deprivation contribute to ferroptosis, and targeting mitochondrial metabolism can reduce these effects and ferroptotic cell death (Figure 2) [12].
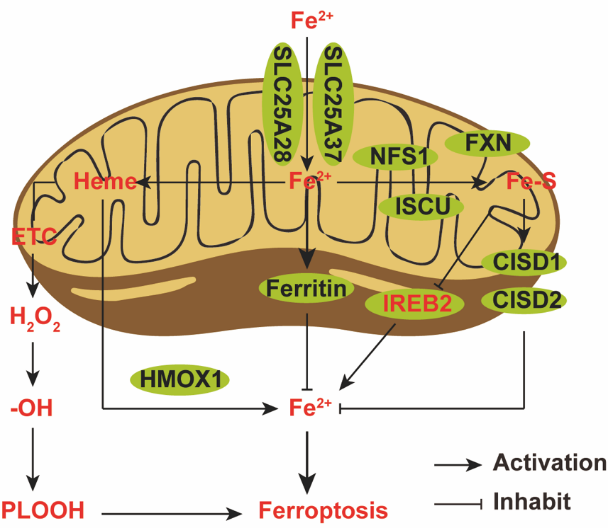
Figure 2. Mitochondrial iron.
3.3. Ferroptosis with Golgi
Various compounds that modulate ferroptosis exhibit protective properties on Golgi structure and function when cells are exposed to the Golgi stressors mentioned earlier. Ferroptosis inhibitors safeguard cells from Golgi dispersion and also impede protein secretion triggered by multiple Golgi stress agents [13].
4. The cellular pathways linking ferroptosis and apoptosis
Ferroptosis is a newly characterized form of regulated cell death uniquely dependent on iron-mediated lipid peroxidation. Its distinguishing characteristic is the triggering of cell death through iron-catalyzed oxidation of lipid molecules. A defining feature of ferroptosis is the accumulation of lipid peroxides and destructive reactive oxygen species. This iron-catalyzed lipid peroxidation is what sets ferroptosis apart as a distinct mode of programmed cell death. The presence of these oxidized lipids and free radicals is a distinguishing characteristic. Extensive research has been conducted on ferroptosis in recent years. Initially, ferroptosis and apoptosis were thought to be mutually exclusive cell death pathways. However, recent evidence suggests that there is significant interplay between ferroptosis and apoptosis. In specific cellular scenarios, these two cellular demise pathways engage in a finely tuned interplay. Multiple subcellular locations and signaling molecules play a role in governing both ferroptosis and apoptosis. These sites and molecules act as modules or switches, allowing the cell to decide which death pathway to activate. Overall, the relationship between ferroptosis and apoptosis is complex and context- dependent. A nuanced understanding of how cells navigate between these two death programs will provide insight into both normal physiology and disease pathology [14].
4.1. The pathways of ferroptosis
Ferroptosis is initiated by an excess of reactive oxygen species (ROS) generated as a result of an unbalanced redox reaction, stemming from reduced glutathione synthesis and the inactivity of the enzyme glutathione peroxidase 4 (GPX4) [15]. This deadly process is characterized by buildup of reactive oxygen species in lipids resulting from iron excess, as well as reduction of polyunsaturated fatty acids in the cell membrane. Cancer cells exhibiting hyperactivation of the RAS-RAF-MEK pathway or p53 expression are often more susceptible to this form of cell death. The interaction between iron, lipid peroxides, and membrane polyunsaturated fatty acids fuels the execution of this lethal cascade [16].
4.2. The pathways of apoptosis
Apoptosis, the regulated cell death process, involves two main pathways, namely, the extrinsic and intrinsic pathways, along with an additional pathway involving perforin and granzymes. Each of these pathways requires specific triggering signals to initiate a series of energy-dependent molecular events. Within each pathway, there are distinct initiator caspases (8, 9, 10) responsible for initiating the activation of the executioner caspase-3 [17].
4.3. Interactions between ferroptosis and apoptosis
I will make comparisons and highlight differences between ferroptosis and other forms of regulated cell death, such as apoptosis. Examining the similarities and differences between ferroptosis and other regulated cell death pathways provides insight into this process [18]. Starvation, as well as the administration of gallic acid and exposure to irradiation, lead to an increase in reactive oxygen species (ROS). Subsequently, ROS induces both apoptosis and ferroptosis, particularly through lipid ROS. TRIP-Br1 reduces ROS levels, with its inhibition being facilitated by the PI3K/Akt pathway. PUFAs instigate ferroptosis by generating lipid ROS, and simultaneously, they activate apoptosis through the PPARγ/SDC-1 pathway. GPX4 and glutathione (GSH) counteract lipid-derived reactive oxygen species (ROS). However, these protective mechanisms are thwarted by compounds like danshen (DT) and PEITC, which also have the capacity to induce apoptosis. Additionally, the tumor suppressor protein p53 serves a versatile function by promoting ferroptosis through the suppression of SLC7A11 and by initiating apoptosis through the regulation of pivotal genes, including BAX, APAF-1, PUMA, p53AIP1, PIDD, and NOXA (Figure 3) [18].
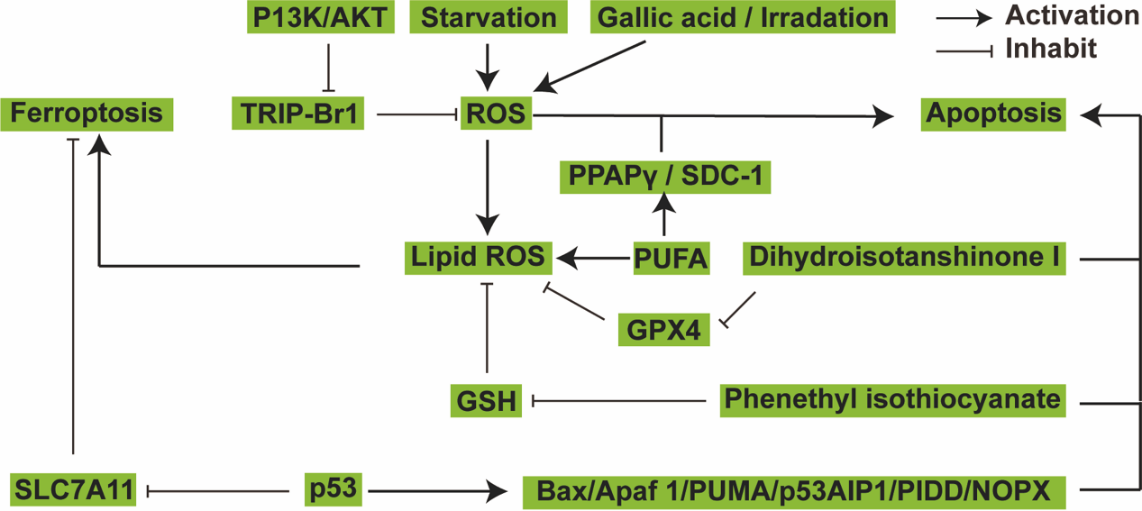
Figure 3. The crosstalk between ferroptosis and apoptosis.
5. Drugs that induce ferroptosis in cells
Stimulating cell death pathways is a prevalent strategy in cancer therapy. Because ferroptosis-triggering compounds can selectively target cancer cells, combining them with current therapies may enhance anti-tumor effects. The cancer cell-specific lethality of ferroptosis inducers makes them promising agents to integrate into existing cytotoxic regimens [4]. Current clinical ferroptosis inducers used for cancer treatment mainly work by inhibiting System Xc- and GPX4 function. The primary anti-tumor mechanism of these compounds involves suppression of these two key proteins. System Xc- is an amino acid transporter that mediates the exchange of extracellular cystine for intracellular glutamate across the plasma membrane. This antiporter activity is essential for the import of cystine and export of glutamate between the outside and inside of the cell (Figure 4) [19]. Moreover, specific natural compounds have a substantial impact on provoking ferroptosis in cancer cells (Table 1).
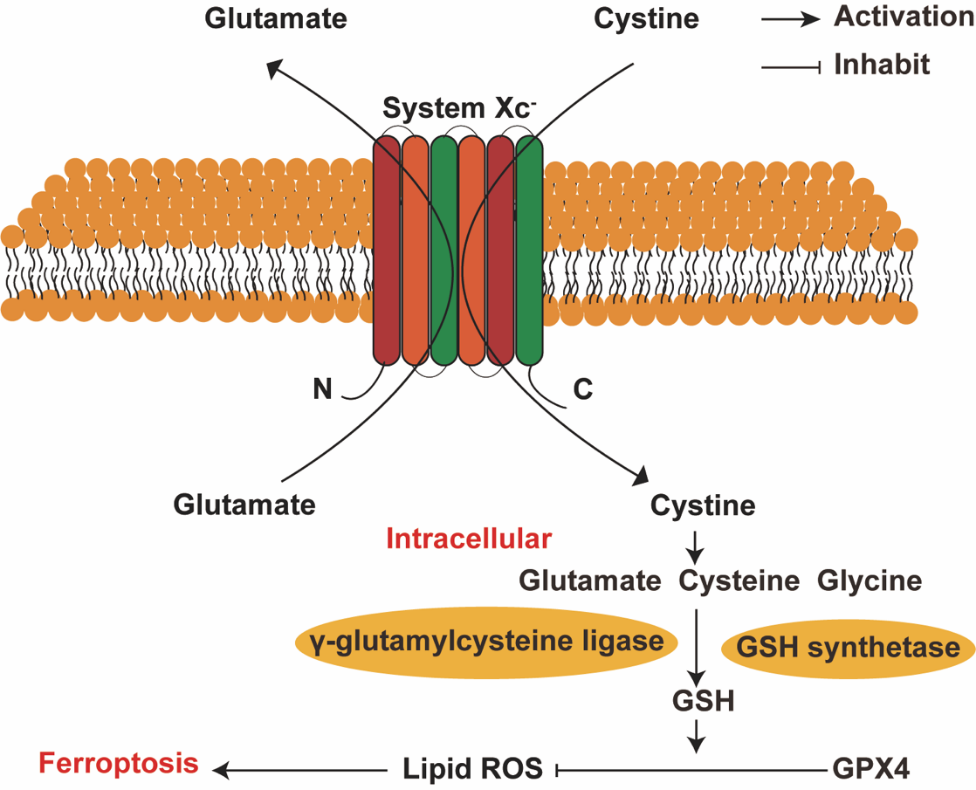
Figure 4. System Xc- Process of transferring amino acid.
Table 1. Preclinical drugs associated with ferroptosis.
Ferroptosis inducer | Clinical/preclinical findings | Mechanisms of action | |
Erastin | Gastric cancer treatment | Inhibit System Xc-, GSH depletion | [4, 20] |
Sulfasalazine | Ovarian cancer and breast cancer treatment | Inhibit System Xc-, GSH depletion | [21] |
Sorafenib | Hepatocellular carcinoma and lung adenocarcinoma | Inhibit System Xc-, GSH depletion | [22] |
RSL3 | Ovarian cancer | GPX4 inactivation | [23, 24] |
ML162 | Induced head and neck cancer cell death | Inhibit GPX4, induces lipid peroxidation | [25] |
ML210 | Mesenchymal-high cancers | Covalently modified selenocysteine of GPX4 | [25] |
FIN56 | Breast carcinoma cells | Deplete CoQ1o, decreases GPX4 level | [5, 26] |
Buthionine sulfoximine (BSO) | Pancreatic cancer | Decreases GSH and GPX4 level | [27] |
Dihydroartemisinin | Glioma cells | Induces ROS, decreases GSH and GPX4 level | [28, 29] |
Siramesine | Breast cancer | Increasing the levels of transferrin and ROS | [30, 31] |
5.1. Inhibition of the System Xc- activity
System Xc- works mainly by swapping intracellular glutamate for extracellular cystine. It moves glutamate out of the cell and brings cystine in from the external environment. This exchange of internal glutamate for external cystine represents the key functional activity of system Xc-. The imported cystine can then be utilized to generate glutathione within the cell. When ferroptosis inducers inhibit System Xc-, the synthesis of glutathione diminishes. Consequently, GPX4 becomes unable to utilize glutathione for the reduction of lipid peroxides, ultimately leading to cell ferroptosis [32]. The primary regulation of System Xc- activity is governed by SCL7A11. A reduction in the expression of SCL7A11 results in decreased System Xc- activity, leading to oxidative stress-induced ferroptosis. Conversely, the upregulation of SCL7A11 enhances the cellular resilience to ferroptosis, a substantial factor contributing to drug resistance in cancer cells [33]. For examples, such drugs are Erastin, Sulfasalazine (SAS) and Sorafenib, etc.
5.1.1. Erastin. Erastin, a small molecule, can trigger ferroptotic cell death in cancer cells, demonstrating significant promise for cancer therapy. However, it is crucial to highlight that erastin-induced ferroptosis led to pathological changes in normal tissues in mouse models as well. This observation implies that the anti-tumor drug erastin may exhibit some level of toxicity to healthy tissues [20]. Erastin has the capability to both prevent and reverse the obstruction of VDAC (voltage-dependent anion channel) by cytoplasmic free tubulin, both in vivo and in vitro, thereby facilitating the opening of the voltage-dependent anion channel [4].
5.1.2. Sulfasalazine. Sulfasalazine (SAS) triggers ferroptosis and exhibits potential in tumor therapy. Glioma cells become more resistant to SAS-induced ferroptosis in low oxygen conditions. The SLC7A11 gene is critical for glioma cells to defend against ferroptosis under hypoxia. Testing the HIF-1α inhibitor PX-478 together with SAS in mouse models revealed it can decrease SLC7A11 levels in tumors, potentiating SAS’s anti-cancer activity. Hypoxic tumor environments limit ferroptosis sensitivity through SLC7A11 upregulation, and inhibiting HIF-1α with PX-478 counters this resistance mechanism [21].
5.1.3. Sorafenib. Cells demonstrating resistance to sorafenib display a marked elevation in intracellular iron content and ROS. Significantly boosting the efficacy of sorafenib as an anti-tumor agent, the suppression of MT-1G expression induces ferroptosis, demonstrating its impact in both in vivo and in vitro settings. Likewise, decreasing expression of ABCC5 substantially improves the anti-cancer efficacy of sorafenib, enhancing its ability to trigger ferroptosis in both cell culture and animal models. Suppressing ABCC5 levels sensitizes tumor cells to sorafenib-induced ferroptosis, underscoring the importance of ABCC5 as a resistance factor that impedes ferroptosis [22].
5.2. Inhibition of the GPX4 activity
Some ferroptosis-stimulating agents act intrinsically to inhibit the antioxidant enzyme GPX4. These compounds promote ferroptosis by directly targeting and suppressing GPX4 activity through internal pathways within the cell [32]. For examples, such drugs are RSL3, ML162, ML210, FIN56 and Buthionine sulfoximine (BSO), etc.
5.2.1. RSL3. RSL3 induces ferroptosis by elevating reactive oxygen species and labile iron levels in cells. RSL3-treated colorectal cancer cells show increased transferrin expression but decreased GPX4, reflecting the iron dependence of this death process. RSL3 drives ferroptosis through iron accumulation combined with oxidative stress and suppression of the lipid antioxidant GPX4 [23]. The growth-inhibitory impact of RSL3 on glioma cells is linked to the disruption of glycolysis and the initiation of autophagy [24].
5.2.2. ML162 and ML210. In both the MDA-MB-231 breast cancer cell lines and Hec50 uterine serous carcinoma cells, the knockout of metadherin (MTDH) resulted in a marked decrease in their responsiveness to GPx4 inhibitors, specifically ML162 and ML210 [25].
5.2.3. FIN56. As a newly identified ferroptosis inducer, FIN56 displayed a marked anti-tumor impact, leading to lysosomal membrane permeabilization through a mechanism that relies on the transcription factor EB (TFEB) in glioma [26].
5.2.4. Buthionine sulfoximine. The influence of L-BSO was evaluated in an in vivo ovarian cancer model, leading to a substantial decrease in subcutaneous tumor size, as well as a reduction in GSH levels and a mitigation of peritoneal dissemination. However, the observed toxicity of L-BSO may limit its suitability for therapeutic use [27].
5.3. Other drugs induce ferroptosis
Dihydroartemisinin (DAT) can stimulate the lysosomal breakdown of ferritin, a process independent of autophagy. This leads to an augmentation of cellular free iron levels, making the cells more vulnerable to ferroptosis [34]. Co-administration of siramesine, a lysosome-destabilizing compound, and the dual tyrosine kinase inhibitor lapatinib synergistically induces cancer cell death in breast tumors. This synergistic lethal effect is achieved predominantly through the induction of ferroptosis. The concurrent lysosomal perturbation caused by siramesine and tyrosine kinase inhibition by lapatinib collaboratively activate ferroptotic cell death pathways in breast cancer cells. The synergistic interplay between these two agents potently promotes ferroptotic cell demise [35]. Inhibition of mitochondrial complex I activity by BAY 87-2243 triggers apoptotic cell death in BRAFV600E mutant melanoma cell lines and impedes melanoma tumor growth in vivo [36].
5.3.1. Dihydroartemisinin. The lethal impact of dihydroartemisinin (DHA) on glioblastoma encompasses several facets, including cytotoxicity, cell cycle arrest, and the suppression of invasion. Artemisinin and its derivatives trigger ferroptosis, resulting in the suppression of head and neck carcinoma and fibrosarcoma [28]. Exposure to DHA detrimentally affects embryos in both rodent and primate models in vitro, primarily through mechanisms that influence cytoskeletal dynamics, oxidative stress, calcium balance, and apoptosis [29].
5.3.2. Siramesine. Siramesine was discovered to have a direct destabilizing effect on the lysosomal membrane. This disruption causes a disturbance in lysosomal operation, ultimately culminating in membrane permeability and the discharge of cathepsins into the cytosol. Consequently, this process results in cell death mediated by cathepsins [30]. Research has shown that Siramesine works synergistically with other agents to elicit ferroptosis, a form of regulated cell death, in breast cancer cells. The mechanism involves Siramesine increasing transferrin levels and promoting ROS accumulation, leading to iron-dependent lipid peroxidation and eventual cell death [31].
6. Potential therapy of cancer through ferroptosis
Immunotherapy employing immune checkpoint inhibitors has marked a significant advancement in oncology and demonstrated considerable effectiveness against various malignancies. Nonetheless, the persistence of drug resistance poses a formidable challenge. The factors contributing to resistance to immunotherapy encompass both inherent characteristics within tumor cells and external elements associated with the tumor microenvironment [37]. Tumor cells’ intrinsic factors, including their genetic or phenotypic traits, can impede the infiltration or proper functioning of immune cells within the tumor microenvironment. In addition to intrinsic factors, extrinsic elements stemming from the tumor microenvironment, excluding the tumor cells themselves, can also suppress anti-tumor immunity [37].
6.1. Tumor-cell-intrinsic mechanisms
Multiple intrinsic properties of tumor cells that confer resistance to immunotherapy have been discovered. These include activation of MAPK signaling, loss of PTEN expression, activation of WNT/β-catenin signaling, defects in interferon-gamma signaling, and downregulation of tumor antigen expression [37]. Inducing ferroptosis, a controlled cell death process, within cancer cells has the potential to generate an immune response akin to vaccination, enhancing anti-tumor immunity and potentially surmounting resistance to immunotherapy [1].
6.2. Tumor-cell-extrinsic mechanisms
Aside from intrinsic tumor cell factors, immunosuppressive cells residing in the tumor microenvironment (TME) can also facilitate resistance to immunotherapy. These immunosuppressive cells encompass regulatory T cells (Tregs) and tumor-associated macrophages (TAMs), both capable of suppressing anti-tumor immunity. The presence of Tregs and TAMs can restrain immune responses against tumors. The presence of these cells in the TME creates an immunosuppressive milieu that enables tumors to evade immune attack [37]. Inducing ferroptosis within the TME could potentially serve as an extrinsic strategy to overcome immunotherapy resistance by eliminating immunosuppressive cells like Tregs and TAMs. Triggering regulated cell death through ferroptosis may therefore deplete these inhibitory immune populations in the TME and enhance anti-tumor immunity.
7. Conclusion
Ferroptosis is a regulated mode of cell death distinct from apoptosis, primarily driven by iron-mediated peroxidation of phospholipids. It is characterized by buildup of ROS and excessive formation of lipid peroxides. The defining features of ferroptosis are iron-catalyzed lipid oxidation leading to ROS accumulation and lipid peroxide overproduction, separating it as a unique form of programmed cell death apart from apoptosis [1]. Regulating cell demise pathways is a traditional tactic in oncology, and ferroptosis specifically has gained recognition as an important component of anti-cancer therapies in modern times. The cellular machinery responsible for regulating ferroptosis assimilates a spectrum of pro-survival and pro-death cues from subcellular organelles, ultimately reaching a verdict on whether to activate this lethal pathway or not [3]. Numerous investigations have showcased the clinical promise of specific ferroptosis inducers. These findings have been pivotal in propelling novel therapeutic approaches for cancer treatment forward. These approaches leverage ferroptosis, a type of iron-reliant programmed cell death, to specifically target and eradicate cancer cells. By elucidating and controlling the mechanisms of ferroptosis, novel treatments seek to activate this cell death process selectively in tumor cells as an anti-cancer tactic. The goal is to exploit the ferroptotic machinery to induce cancer cell demise in a precise manner [4].
In this review, this summarizes the current comprehension of the regulatory mechanisms that govern ferroptosis, the interplay between apoptosis and ferroptosis, as well as the association between ferroptosis and specific cellular organelles. Finally, some examples of drugs related to this. While some inducers of ferroptosis are currently being explored in clinical trials, clinical data remains insufficient at this time. There are presently no approved drugs leveraging ferroptotic cell death mechanisms available on the market, and significant work remains to translate our understanding of iron-dependent cell death pathways into practical clinical applications.
References
[1]. Zhang C, Liu X, Jin S, Chen Y and Guo R 2022 Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer, 21(1) 47.
[2]. Dietrich C and Hofmann T G 2021 Ferroptosis Meets Cell-Cell Contacts. Cells, 10(9).
[3]. Chen X, Kang R, Kroemer G and Tang D 2021 Organelle-specific regulation of ferroptosis. Cell Death Differ, 28(10) 2843-56.
[4]. Zhao Y, Li Y, Zhang R, Wang F, Wang T and Jiao Y 2020 The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther, 13 5429-41.
[5]. Tang D, Chen X, Kang R and Kroemer G 2021 Ferroptosis: molecular mechanisms and health implications. Cell Res, 31(2) 107-25.
[6]. Li J, Cao F, Yin H L, Huang Z J, Lin Z T, Mao N, Sun B and Wang G 2020 Ferroptosis: past, present and future. Cell Death Dis, 11(2) 88.
[7]. Qu L, He X, Tang Q, Fan X, Liu J and Lin A 2022 Iron metabolism, ferroptosis, and lncRNA in cancer: knowns and unknowns. J Zhejiang Univ Sci B, 23(10) 844-62.
[8]. Yan H F, Zou T, Tuo Q Z, Xu S, Li H, Belaidi A A and Lei P 2021 Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther, 6(1) 49.
[9]. Song W, Zhang W, Yue L and Lin W 2022 Revealing the Effects of Endoplasmic Reticulum Stress on Ferroptosis by Two-Channel Real-Time Imaging of pH and Viscosity. Anal Chem, 94(17) 6557-65.
[10]. Silswal A and Koner A L 2023 Tracking endoplasmic reticulum viscosity during ferroptosis and autophagy using a molecular rotor probe. Chem Commun (Camb), 59(13) 1769-72.
[11]. Yang Z, Cao J, He Y, Yang J H, Kim T, Peng X and Kim J S 2014 Macro-/micro-environment-sensitive chemosensing and biological imaging. Chem Soc Rev, 43(13) 4563-601.
[12]. Gao M, Yi J, Zhu J, Minikes A M, Monian P, Thompson C B and Jiang X 2019 Role of Mitochondria in Ferroptosis. Mol Cell, 73(2) 354-63 e3.
[13]. Garg S K, Yan Z, Vitvitsky V and Banerjee R 2011 Differential dependence on cysteine from transsulfuration versus transport during T cell activation. Antioxid Redox Signal, 15(1) 39-47.
[14]. Yang J, Hu S, Bian Y, Yao J, Wang D, Liu X, Guo Z, Zhang S and Peng L 2021 Targeting Cell Death: Pyroptosis, Ferroptosis, Apoptosis and Necroptosis in Osteoarthritis. Front Cell Dev Biol, 9 789948.
[15]. Li J Y, Yao Y M and Tian Y P 2021 Ferroptosis: A Trigger of Proinflammatory State Progression to Immunogenicity in Necroinflammatory Disease. Front Immunol, 12 701163.
[16]. Cao J Y and Dixon S J 2016 Mechanisms of ferroptosis. Cell Mol Life Sci, 73(11-12) 2195-209.
[17]. Elmore S 2007 Apoptosis: a review of programmed cell death. Toxicol Pathol, 35(4) 495-516.
[18]. Li Zet al. 2020 Targeting ferroptosis in breast cancer. Biomark Res, 8(1) 58.
[19]. Bridges R J, Natale N R and Patel S A 2012 System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol, 165(1) 20-34.
[20]. Zhao J, Xu B, Xiong Q, Feng Y and Du H 2021 Erastin‑induced ferroptosis causes physiological and pathological changes in healthy tissues of mice. Mol Med Rep, 24(4).
[21]. Sun S, Guo C, Gao T, Ma D, Su X, Pang Q and Zhang R 2022 Hypoxia Enhances Glioma Resistance to Sulfasalazine-Induced Ferroptosis by Upregulating SLC7A11 via PI3K/AKT/HIF-1alpha Axis. Oxid Med Cell Longev, 2022 7862430.
[22]. Guo L, Hu C, Yao M and Han G 2023 Mechanism of sorafenib resistance associated with ferroptosis in HCC. Front Pharmacol, 14 1207496.
[23]. Sui Xet al. 2018 RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol, 9 1371.
[24]. Wang X, Lu S, He C, Wang C, Wang L, Piao M, Chi G, Luo Y and Ge P 2019 RSL3 induced autophagic death in glioma cells via causing glycolysis dysfunction. Biochem Biophys Res Commun, 518(3) 590-7.
[25]. Bi Jet al. 2019 Metadherin enhances vulnerability of cancer cells to ferroptosis. Cell Death Dis, 10(10) 682.
[26]. Zhang X, Guo Y, Li H and Han L 2021 FIN56, a novel ferroptosis inducer, triggers lysosomal membrane permeabilization in a TFEB-dependent manner in glioblastoma. J Cancer, 12(22) 6610-9.
[27]. Cruz A, Mota P, Ramos C, Pires R F, Mendes C, Silva J P, Nunes S C, Bonifacio V D B and Serpa J 2020 Polyurea Dendrimer Folate-Targeted Nanodelivery of l-Buthionine sulfoximine as a Tool to Tackle Ovarian Cancer Chemoresistance. Antioxidants (Basel), 9(2).
[28]. Yi R, Wang H, Deng C, Wang X, Yao L, Niu W, Fei M and Zhaba W 2020 Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Biosci Rep, 40(6).
[29]. Luo Y, Che M J, Liu C, Liu H G, Fu X W and Hou Y P 2018 Toxicity and related mechanisms of dihydroartemisinin on porcine oocyte maturation in vitro. Toxicol Appl Pharmacol, 341 8-15.
[30]. Jensen S S, Petterson S A, Halle B, Aaberg-Jessen C and Kristensen B W 2017 Effects of the lysosomal destabilizing drug siramesine on glioblastoma in vitro and in vivo. BMC Cancer, 17(1) 178.
[31]. Zhao Let al. 2022 Ferroptosis in cancer and cancer immunotherapy. Cancer Commun (Lond), 42(2) 88-116.
[32]. Wang Det al. 2022 Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis, 13(6) 544.
[33]. Li F J, Long H Z, Zhou Z W, Luo H Y, Xu S G and Gao L C 2022 System X(c) (-)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol, 13 910292.
[34]. Chen G Q, Benthani F A, Wu J, Liang D, Bian Z X and Jiang X 2020 Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ, 27(1) 242-54.
[35]. Ma S, Dielschneider R F, Henson E S, Xiao W, Choquette T R, Blankstein A R, Chen Y and Gibson S B 2017 Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One, 12(8) e0182921.
[36]. Basit Fet al. 2017 Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis, 8(3) e2716.
[37]. Sharma P, Hu-Lieskovan S, Wargo J A and Ribas A 2017 Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell, 168(4) 707-23.
Cite this article
Yuan,Y. (2023). The prospects of targeting ferroptosis in cancer therapy. Theoretical and Natural Science,27,148-157.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Zhang C, Liu X, Jin S, Chen Y and Guo R 2022 Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer, 21(1) 47.
[2]. Dietrich C and Hofmann T G 2021 Ferroptosis Meets Cell-Cell Contacts. Cells, 10(9).
[3]. Chen X, Kang R, Kroemer G and Tang D 2021 Organelle-specific regulation of ferroptosis. Cell Death Differ, 28(10) 2843-56.
[4]. Zhao Y, Li Y, Zhang R, Wang F, Wang T and Jiao Y 2020 The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther, 13 5429-41.
[5]. Tang D, Chen X, Kang R and Kroemer G 2021 Ferroptosis: molecular mechanisms and health implications. Cell Res, 31(2) 107-25.
[6]. Li J, Cao F, Yin H L, Huang Z J, Lin Z T, Mao N, Sun B and Wang G 2020 Ferroptosis: past, present and future. Cell Death Dis, 11(2) 88.
[7]. Qu L, He X, Tang Q, Fan X, Liu J and Lin A 2022 Iron metabolism, ferroptosis, and lncRNA in cancer: knowns and unknowns. J Zhejiang Univ Sci B, 23(10) 844-62.
[8]. Yan H F, Zou T, Tuo Q Z, Xu S, Li H, Belaidi A A and Lei P 2021 Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther, 6(1) 49.
[9]. Song W, Zhang W, Yue L and Lin W 2022 Revealing the Effects of Endoplasmic Reticulum Stress on Ferroptosis by Two-Channel Real-Time Imaging of pH and Viscosity. Anal Chem, 94(17) 6557-65.
[10]. Silswal A and Koner A L 2023 Tracking endoplasmic reticulum viscosity during ferroptosis and autophagy using a molecular rotor probe. Chem Commun (Camb), 59(13) 1769-72.
[11]. Yang Z, Cao J, He Y, Yang J H, Kim T, Peng X and Kim J S 2014 Macro-/micro-environment-sensitive chemosensing and biological imaging. Chem Soc Rev, 43(13) 4563-601.
[12]. Gao M, Yi J, Zhu J, Minikes A M, Monian P, Thompson C B and Jiang X 2019 Role of Mitochondria in Ferroptosis. Mol Cell, 73(2) 354-63 e3.
[13]. Garg S K, Yan Z, Vitvitsky V and Banerjee R 2011 Differential dependence on cysteine from transsulfuration versus transport during T cell activation. Antioxid Redox Signal, 15(1) 39-47.
[14]. Yang J, Hu S, Bian Y, Yao J, Wang D, Liu X, Guo Z, Zhang S and Peng L 2021 Targeting Cell Death: Pyroptosis, Ferroptosis, Apoptosis and Necroptosis in Osteoarthritis. Front Cell Dev Biol, 9 789948.
[15]. Li J Y, Yao Y M and Tian Y P 2021 Ferroptosis: A Trigger of Proinflammatory State Progression to Immunogenicity in Necroinflammatory Disease. Front Immunol, 12 701163.
[16]. Cao J Y and Dixon S J 2016 Mechanisms of ferroptosis. Cell Mol Life Sci, 73(11-12) 2195-209.
[17]. Elmore S 2007 Apoptosis: a review of programmed cell death. Toxicol Pathol, 35(4) 495-516.
[18]. Li Zet al. 2020 Targeting ferroptosis in breast cancer. Biomark Res, 8(1) 58.
[19]. Bridges R J, Natale N R and Patel S A 2012 System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol, 165(1) 20-34.
[20]. Zhao J, Xu B, Xiong Q, Feng Y and Du H 2021 Erastin‑induced ferroptosis causes physiological and pathological changes in healthy tissues of mice. Mol Med Rep, 24(4).
[21]. Sun S, Guo C, Gao T, Ma D, Su X, Pang Q and Zhang R 2022 Hypoxia Enhances Glioma Resistance to Sulfasalazine-Induced Ferroptosis by Upregulating SLC7A11 via PI3K/AKT/HIF-1alpha Axis. Oxid Med Cell Longev, 2022 7862430.
[22]. Guo L, Hu C, Yao M and Han G 2023 Mechanism of sorafenib resistance associated with ferroptosis in HCC. Front Pharmacol, 14 1207496.
[23]. Sui Xet al. 2018 RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol, 9 1371.
[24]. Wang X, Lu S, He C, Wang C, Wang L, Piao M, Chi G, Luo Y and Ge P 2019 RSL3 induced autophagic death in glioma cells via causing glycolysis dysfunction. Biochem Biophys Res Commun, 518(3) 590-7.
[25]. Bi Jet al. 2019 Metadherin enhances vulnerability of cancer cells to ferroptosis. Cell Death Dis, 10(10) 682.
[26]. Zhang X, Guo Y, Li H and Han L 2021 FIN56, a novel ferroptosis inducer, triggers lysosomal membrane permeabilization in a TFEB-dependent manner in glioblastoma. J Cancer, 12(22) 6610-9.
[27]. Cruz A, Mota P, Ramos C, Pires R F, Mendes C, Silva J P, Nunes S C, Bonifacio V D B and Serpa J 2020 Polyurea Dendrimer Folate-Targeted Nanodelivery of l-Buthionine sulfoximine as a Tool to Tackle Ovarian Cancer Chemoresistance. Antioxidants (Basel), 9(2).
[28]. Yi R, Wang H, Deng C, Wang X, Yao L, Niu W, Fei M and Zhaba W 2020 Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Biosci Rep, 40(6).
[29]. Luo Y, Che M J, Liu C, Liu H G, Fu X W and Hou Y P 2018 Toxicity and related mechanisms of dihydroartemisinin on porcine oocyte maturation in vitro. Toxicol Appl Pharmacol, 341 8-15.
[30]. Jensen S S, Petterson S A, Halle B, Aaberg-Jessen C and Kristensen B W 2017 Effects of the lysosomal destabilizing drug siramesine on glioblastoma in vitro and in vivo. BMC Cancer, 17(1) 178.
[31]. Zhao Let al. 2022 Ferroptosis in cancer and cancer immunotherapy. Cancer Commun (Lond), 42(2) 88-116.
[32]. Wang Det al. 2022 Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis, 13(6) 544.
[33]. Li F J, Long H Z, Zhou Z W, Luo H Y, Xu S G and Gao L C 2022 System X(c) (-)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol, 13 910292.
[34]. Chen G Q, Benthani F A, Wu J, Liang D, Bian Z X and Jiang X 2020 Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ, 27(1) 242-54.
[35]. Ma S, Dielschneider R F, Henson E S, Xiao W, Choquette T R, Blankstein A R, Chen Y and Gibson S B 2017 Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One, 12(8) e0182921.
[36]. Basit Fet al. 2017 Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis, 8(3) e2716.
[37]. Sharma P, Hu-Lieskovan S, Wargo J A and Ribas A 2017 Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell, 168(4) 707-23.