







1. Introduction
The study of heritable differences in gene expression or function that are not caused by changes in the sequence of the deoxyribonucleic acid (DNA) is known as Epigenetics. Not all types of cells will have the same gene expression at once, even though an organism’s cells nearly all share the same genetic materials. Epigenetic mechanisms, in a larger sense, regulate the varied patterns of gene expression in many different kinds of tissues and cell types in multicellular creatures. DNA methylation was first identified in mammals when DNA was first recognized as the genetic material. Using paper chromatography, Rollin Hotchkiss made the initial discovery of modified cytosine in a calf thymus sample in 1948 [1].
DNA methyltransferases (DNMTs) covalently add a methyl group to the C-5 position of the DNA cytosine ring, resulting in DNA methylation, an epigenetic alteration. In somatic cells, more than 98% of DNA methylation occurs in a CpG dinucleotide environment, nevertheless in embryonic stem cells (ESCs), up to 25% of total DNA methylation occurs in non-CpG settings. Most DNA methylation is essential for several important activities, including X-chromosome inactivation and genomic imprinting. DNA methylation can also be improper and lead to illnesses like cancer [2]. The methylation of DNA is regulated by a family of DNMTs, including DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L. The majority of CG dinucleotides in mammals are methylated on cytosine residues, although CG dinucleotides found in promoters are typically shielded from methylation. Global DNA hypomethylation and locus-specific CpG island (CGI) hypermethylation are epigenetic indicators of malignancy. All of the cancer specimens that have been analyzed so far exhibit overall decreases in DNA methylation. The primary underlying cause of hypomethylation is a lack of methylation at repetitive elements that are typically strongly methylated, such as satellites and retrotransposons, which results in genomic unpredictability and oncogene stimulation. Typically, suppressive tumor genes’ promoter CGIs experience locus-specific hypermethylation, which causes transmissible transcriptional silence [2].
One of the most significant inventions that helps scientists identify the patterns of methylation is the development of DNA sequencing. DNA sequencing implies to a common analytical procedure for figuring out the specific arrangement of nucleotides, also called bases, in a DNA molecule. The biological information that living organisms use for development and function is stored in the DNA sequence. Identifying the roles of genes and other components of the genome has become achievable as a consequence to the advancement in the availability of genetic information in our genomes made possible by DNA sequencing [3].
This review described three different serious diseases whose causes have been studied to be related to DNA methylation. Moreover, it introduced two different sequencing techniques utilized to analyze DNA information for identifying methylation patterns. By utilizing these advanced DNA sequencing techniques helps scientists to investigate the genetic-level causations of serious diseases, which provides potential effective treatments for these diseases.
2. DNA methylation-linked diseases
Mammalian embryogenesis and optimal adult organism operation depend on appropriately created and retained DNA methylation patterns. DNA methylation is a powerful method for suppressing gene expression and maintaining genomic integrity in the face of this abundance of repetitive DNA. Dnmt1 and Dnmt3b are required for the development of embryos, and mice lacking Dnmt3a passed away shortly after birth, according to research studies of mouse knockout. Additionally, the somatic cells lose the ability to control their proliferation when their DNA methylation patterns revert to normal. The prevalence of human diseases emphasizes the significance of DNA methylation [4]. The following section presents the research findings of three different diseases.
2.1. Breast cancer
The most common hormone-dependent malignancy that causes a high rate of mortality as well as morbidity is carcinoma of the breast. This frequent cancer begins as a hormone-responsive (HR), non-metastatic cancer throughout the intricate, multi-step process of tumor promotion, then gradually transforms into a highly metastatic hormone-insensitive (HI) version that lacks the functioning estrogen receptor. Several malignancies, including breast cancer, have been linked to the progression of urokinase (uPA), a member of the serine protease family. In this study, scientists have investigated the relationship between uPA expression and hormone sensitivity in HR normal mammary epithelial cells, MCF-7, and T-47D breast cancer cell lines [5].
To performing experiments, researchers utilized human mammary epithelial cell line (HMEC). In order to attain the final results, researchers utilized northern blot analysis, nuclear run-off assay, Matrigel invasion assay, and southern blot analysis [5].
DNA from the genome was extracted from HR and HI mammary epithelial cells with the aim of investigating the significance of the methylation of DNA in the variation in the regulation of control of uPA gene activity throughout cancer metamorphosis and tumor development. Total cellular DNA was digested using either Pst 1, a non-methylation-sensitive endonuclease, only (Fig. 2; lanes 1, 4, 7, and 10), or Pst 1 combined with either Hpa11 or Hha1, a methylation-sensitive endonuclease (lanes 2, 5, 8, and 11). Southern blotting was used to determine whether the Hpa11 and Hha1 sites were methylated. A 778 bp Sma I and Avr II promoter region within the uPA gene was used to probe all blots. In HR HMEC, MCF-7, and T-47D cells, methylating the uPA gene’s CpG island prevented DNA from being digested by Hpa11 and Hha1, resulting in patterns of restriction that were the same as those of Pst 1 digestion alone (Fig. 1; lanes 1–9). Hpa11 and Hha1 completely digested the genomic DNA to produce many lower bands as opposed to Pst1 digestion alone in HI MDA-231 cells when the CpG island of the uPA gene was methylation or hypomethylated (Fig. 1; lanes 10–12) [5].
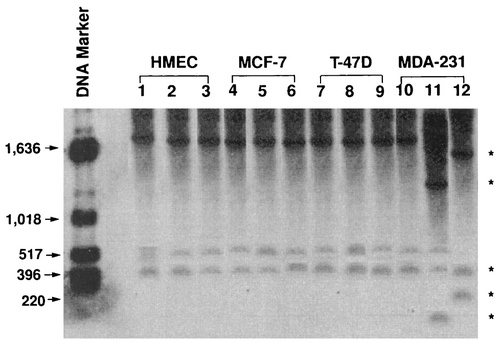
Figure 1. Methylation status of key sites on the human uPA gene’s CpG island. Genomic DNA isolated from HR (HMEC, MCF-7, T-47D) and H1 (MDA-231) mammary epithelial cells [5].
Their results provide compelling evidence for an epigenetic pathway involving the methylation of DNA that regulates the transcription-dependent control of uPA gene expression and allows for the on and off regulation of this vital protease at various phases of breast cancer.
2.2. Autism spectrum disorder
A series of neurodevelopmental diseases known as autism spectrum disorder (ASD) are characterized by impaired social interaction and communication, as well as more frequent repetitive activities and narrowed interests. All races, all ethnic groups, and all socioeconomic categories are affected by the condition, which affects males more commonly than females (by a ratio of 4.5 to 1). Even if the majority of ASD cases are not inherited, the substantial consistency of ASD among identical twins points to a significant genetic link as well as complicated transmission. The etiologic involvement of genetic flaws in ASD has also been convincingly demonstrated by genomic findings during the previous two decades [6]. In this research, scientists employed nuclease-deactivated Cas9 (dCas9) coupled with DNA methyltransferase catalytic domains and five locus-targeting sgRNAs to accomplish locus-specific methylation at the location of the transcription start site (TSS) of Mecp2 in Neuro-2a cells and mice. They investigated the connection between ASD symptoms and locus-specific DNA methylation [7].
To get the most accurate results, the researcher utilized plenty of different techniques during the experiments, including T7ENI cleavage assay and sequencing, immunofluorescent staining, behavioral assessments pertaining to motor skills, anxiety, and depression, and reduced representation bisulfite sequencing [7].
After researchers methylated the promoter of Mecp2, they discovered that male mouse from the experimental methylation collections had considerably less Mecp2 transcription in the hippocampus and parietal cortex when compared to their control group. Additionally, in contrast to masculine mouse serving as controls, the MeCP2 levels in tissue sections were much reduced as shown by a Western blot analysis, which was supported by a weak immunofluorescence labeling of the MeCP2 protein (Fig. 2D, E) [7].
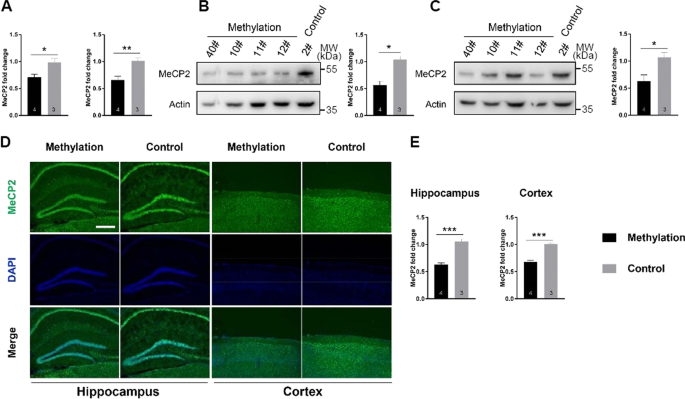
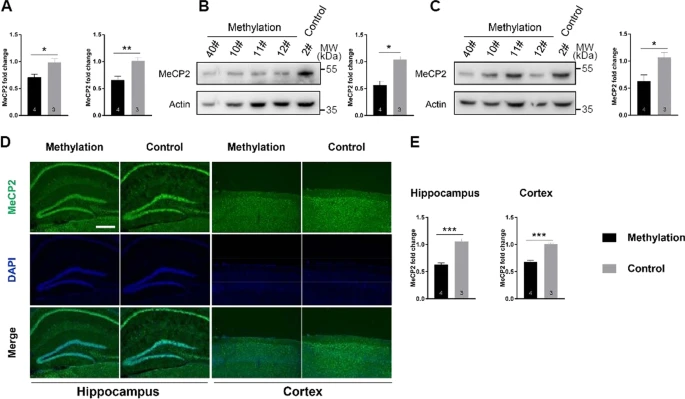
Figure 2. Reduced expression of Mecp2’s by targeted DNA methylation [7].
Then, researchers examined whether changing the DNA methylation at the Mecp2 promoter would result in altered mouse behavior. The interpersonal communication test consisted of three separate chambers. When confronted with unknown mouse, mouse in the untreated group preferred to investigate them instead of known mice, while mice in the methylation sample showed no preference between known and unknown mice (Fig 3B). Additionally, we discovered that mouse from the methylation collections spent noticeably more time grooming themselves (Fig. 3C), which indicates an increase in repetitive behavior [7].
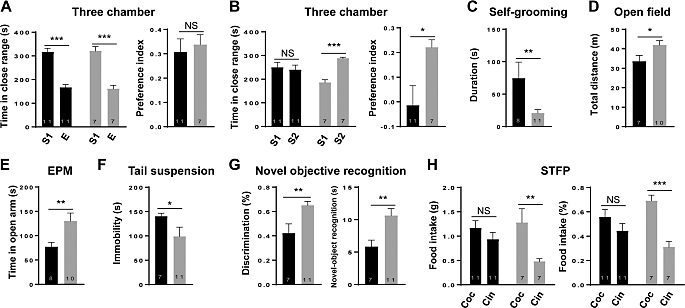
Figure 3. Mecp2’s targeted DNA methylation in mice resulted in characteristics similar to ASD [7].
By adopting an enhanced dCas9-based methylation targeting approach to methylate Mecp2 promotor at a specific location, scientists were able to illustrate the crucial role that methylation plays in the pathophysiology of ASD. Mecp2 gene expression was decreased as a result of methylating the Mecp2 promotor. Additionally, the data revealed that methylation group animals were less interested in social novelty or conspecific recognition than control mice.
2.3. Obesity
Over the past few years, childhood obesity prevalence has grown significantly and quickly, turning it into a global public health emergency. In order to determine the methylation biomarkers in obesity, researchers investigated into genome-wide methylation of the DNA in this study. In order to identify the distinctively methylated genes, they used extensive DNA methylation characterization of promoters of genes and CpG islands in obese Chinese preschoolers. This was followed by methylated DNA immunoprecipitation and hybridization to the NimbleGen Human DNA Methylation 385K Promoter Plus CpG Island Microarray [8]. In the study, 748 children from kindergartens in Beijing, including 32 obese preschoolers aged 3 to 6 and an equal number of lean (normal weight) youngsters who were age- and gender-matched, were included. Scientists utilized immunoprecipitation, DNA labeling and array hybridization, and sequencing of bisulfite-modified DNA to obtain their final results [8].
In comparison to lean children, 251 promoters were demethylated in the peripheral blood cells of obese kids, while 141 were hypermethylated. These promoters were connected to a total of 232 and 132 genes, respectively. In addition, when comparing obese children to control group youngsters, 575 CpG islands were demethylated and 277 were hypermethylated (Fig. 4). Further investigation revealed that their chromosomal distribution was unbalanced, particularly on chromosomes 3, 16, 17 and 19, which contained more promoters and demethylated CpG islands. Additionally, it was discovered that chromosome X had more CpG islands and promoters with differential methylation than Y chromosome [8].
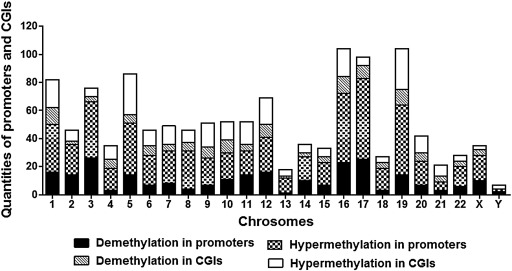
Figure 4. Distribution of CpG islands (CGIs) and promoters with varied levels of methylation in obese children’s chromosomes [8].
Furthermore, of the 80 most significant promoters and CGIs with DNA methylation distinctions among control and obese children, almost fifty percent have been studied previously, and the majority of these are associated with the etiology of malignancies that are connected to multiple body parts, including glioblastoma, musculoaponeurotic fibrosarcoma, lung carcinoma, leukemia, and malignant melanoma [8].
3. DNA methylation-linked analytical methods
DNA sequencing played a very crucial role in mapping out the human genome, and it’s still a significant tool for both fundamental and applied scientific applications today. As an example, it has provided an essential instrument for determining the numerous numbers of nucleotide variations connected to specific genetic diseases, which may aid in improving understanding of these conditions and therapy. Following the discovery of the mechanisms underlying methylation, scientists came upon sequencing analysis that was specifically utilized to identify the methylation patterns of CpG, CHH, and CHG sites throughout the human genome [9]. The following section introduces two different sequencing techniques that are very helpful for researchers to conduct their studies about methylation.
3.1. Bisulfite conversion-based assays
The gold-standard technique: DNA bisulfite conversion followed by sequencing is used to quantify patterns of methylation at the base-pair level. Both on microarrays and by NextGen sequencing, the first step in assessing DNA methylation can be bisulfite conversion-based methods [10]. Frommer et al. first introduced the bisulfite treatment in 1992 [11].
Researchers utilize the bisulfite sequencing to investigate 5-methylcytosine (5mC) in the single strands sequence. Unmethylated cytosines are converted to uracil by the bisulfite process, causing U to be read as T following PCR amplification and sequencing. Methylated cytosines are unaffected by this change and remain C in the sequence (Fig. 5). To determine the status of DNA methylation, PCR amplicons created following DNA from the genome that has undergone bisulfite conversion can be matched to microarrays containing methylation-specific oligonucleotides (MSOs), which are composed of 19–23 nucleotides. The fluorescence intensity determines the extent of the variations in hybridization, might provide information on the methylation state of a specific locus since MSO probes could distinguish between methylated and unmethylated cytosines within a given CG-rich sequence. Additional research using MSO allowed for the classification of different human tumor types according to methylation patterns. Studying colorectal cancer can be achieved by a modified version of MSO technology that shows the methylation status of the MGMT gene’s promoter region [10].
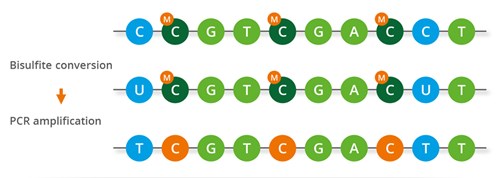
Figure 5. The mechanisms of Bisulfite conversion-based assays [12].
3.2. Enzymatic methyl sequencing
At single-base resolution, 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) are found using bisulfite sequencing. However, DNA is harmed by bisulfite treatment, leading to DNA loss, fragmentation, and inaccurate sequencing results. Enzymatic methyl-seq (EM-seq), an fully enzyme-based technique for assessing the methylation state of cytosine, was created to address these issues [13].
Three enzymes and a pair of reactions are needed for the enzyme-mediated identification of 5mC and 5hmC. T4-BGT and Tet methylcytosine dioxygenase 2 (TET2) are employed to shield 5mC and 5hmC from APOBEC3A’s ensuing deamination. TET2 has the ability to catalyze the oxidation of 5mC to 5hmC, then 5-formylcytosine (5fC) to 5-carboxycytosine (5caC), resulting in simultaneous production of CO2 and succinic acid (Fig. 6A). TET2-formed and genome-wide 5hmC are both glucosylated to 5-(β-glucosyloxymethyl) cytosine (5gmC) by T4-BGT (Fig. 6B). Then, cytosines are deaminated by APOBEC3A (Fig. 6C) [13].
With fewer sequencing duplicates than bisulfite sequencing, EM-seq produced more usable reads, which increased the effective genome coverage.
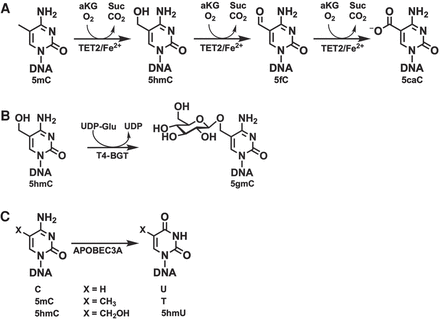
Figure 6. Enzymes that are utilized to detect 5mC and 5hmC [13].
4. Conclusion
Based on the technologies that have been developed to study genetics and genomics, scientists have been studying the reasons behind serious diseases at the genetic level for many years. The DNA/histone modifications of epigenetics have also become one of the most popular research fields in recent years. This overview introduced the background, methods, and results of three diseases with different etiologies related to DNA methylation: breast cancer, autism spectrum disorder, and obesity and two different sequencing methods used to identify DNA methylation patterns: bisulfite conversion-based assays and enzymatic methyl sequencing. Due to the lack of objective data, this overview does not include the data from patients’ clinical trials on autism spectrum disorder that can support the conclusion. With the advanced discoveries and developments of technology, it becomes possible for researchers to study more and more diseases to discover the fundamental causes behind them in the future, and some of their causes might be related to epigenetics. With the knowledge of the causes of diseases, scientists can invent better therapies for treating serious diseases.
References
[1]. Moore, L., Le, Thuc, & Fan, Guoping. (2012). DNA Methylation and Its Basic Function. Neuropsychopharmacology, 38, 23–38. https://doi.org/10.1038/npp.2012.112
[2]. Jin, B., Li, Y., & Robertson, Keith. (2011). DNA Methylation. Genes Cancer, 2(6), 607–617. https://doi.org/10.1177/1947601910393957
[3]. DNA Sequencing. (2023).National Human Genome Research Institute; National Human Genome Research Institute https://doi.org/10.1177/1947601910393957
[4]. Robertson, K. (2005). DNA methylation and human disease. Nature Reviews Genetics, 6, 597–610. https://doi.org/10.1038/nrg1655
[5]. Xing, R., & Rabbani, S. (1999). Transcriptional regulation of urokinase (uPA) gene expression in breast cancer cells: Role of DNA methylation. International Journal of Cancer, 81(3), 443–450. https://doi.org/10.1002/(SICI)1097-0215(19990505)81:3<443::AID-IJC19>3.0.CO;2-T
[6]. Tremblay, M., & Jiang, Y. (2019). DNA Methylation and Susceptibility to Autism Spectrum Disorder. Annu Rev Med, 70, 151–166. https://doi.org/10.1146/annurev-med-120417-091431
[7]. Lu, Z., Liu, Zhen, Mao, Wei, Wang, Xinying, Zheng, X., Chen, S., Cao, B., Huang, S., Zhang, X., Zhou, T., Zhang, Y., Huang, X., Sun, Q., & Li, J.-D. (2020). Locus-specific DNA methylation of Mecp2 promoter leads to autism-like phenotypes in mice. Cell Death & Disease, 11(85). https://doi.org/10.1038/s41419-020-2290-x
[8]. Ding, X., Zheng, D., Fan, C., Liu, Zhaoqiu, Dong, H., Lu, Y., & Qi, K. (2015). Genome-wide screen of DNA methylation identifies novel markers in childhood obesity. Gene, 566(1), 74–83. https://doi.org/10.1016/j.gene.2015.04.032
[9]. Methylation Sequencing. (2023). Illumina; Illumina, Inc. https://doi.org/10.1016/j.gene.2015. 04.032
[10]. Zuo, T., Tycko, B., Liu, T.-M., Lin, H.-J., & Huang, T. (2009). Methods in DNA methylation profiling. Epigenomics, 1(2). https://doi.org/10.2217/epi.09.31
[11]. Frommer, M., McDonald, L., Millar, D., Collist, C., Watt, F., Grigg, G., Molloy, P., & Paul, C. (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Genetics, 89, 1827–1831.
[12]. Whole-genome Bisulfite Sequencing. (2023). Eurofins Genomics; Eurofins Genomics Europe Shared Services GmbH. https://doi.org/10.1101/gr.266551.120
[13]. Vaisvila, R., Ponnaluri, V. K. C., Sun, Z., Langhorst, B., Saleh, L., Guan, S., Dai, Nan, Campbell, Matthew, Sexton, Brittany, Marks, K., Samaranayake, M., Samuelson, J., Church, H., Tamanaha, E., Corrêa Jr., Ivan, Pradhan, S., Dimalanta, Eileen, Evans Jr., T., Williams, L., & Davis, T. (2021). Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res., 31, 1280–1289. https://doi.org/10.1101/gr.266551.120
Cite this article
Gao,Z. (2023). DNA methylation-linked diseases and analytical methods. Theoretical and Natural Science,27,223-229.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Moore, L., Le, Thuc, & Fan, Guoping. (2012). DNA Methylation and Its Basic Function. Neuropsychopharmacology, 38, 23–38. https://doi.org/10.1038/npp.2012.112
[2]. Jin, B., Li, Y., & Robertson, Keith. (2011). DNA Methylation. Genes Cancer, 2(6), 607–617. https://doi.org/10.1177/1947601910393957
[3]. DNA Sequencing. (2023).National Human Genome Research Institute; National Human Genome Research Institute https://doi.org/10.1177/1947601910393957
[4]. Robertson, K. (2005). DNA methylation and human disease. Nature Reviews Genetics, 6, 597–610. https://doi.org/10.1038/nrg1655
[5]. Xing, R., & Rabbani, S. (1999). Transcriptional regulation of urokinase (uPA) gene expression in breast cancer cells: Role of DNA methylation. International Journal of Cancer, 81(3), 443–450. https://doi.org/10.1002/(SICI)1097-0215(19990505)81:3<443::AID-IJC19>3.0.CO;2-T
[6]. Tremblay, M., & Jiang, Y. (2019). DNA Methylation and Susceptibility to Autism Spectrum Disorder. Annu Rev Med, 70, 151–166. https://doi.org/10.1146/annurev-med-120417-091431
[7]. Lu, Z., Liu, Zhen, Mao, Wei, Wang, Xinying, Zheng, X., Chen, S., Cao, B., Huang, S., Zhang, X., Zhou, T., Zhang, Y., Huang, X., Sun, Q., & Li, J.-D. (2020). Locus-specific DNA methylation of Mecp2 promoter leads to autism-like phenotypes in mice. Cell Death & Disease, 11(85). https://doi.org/10.1038/s41419-020-2290-x
[8]. Ding, X., Zheng, D., Fan, C., Liu, Zhaoqiu, Dong, H., Lu, Y., & Qi, K. (2015). Genome-wide screen of DNA methylation identifies novel markers in childhood obesity. Gene, 566(1), 74–83. https://doi.org/10.1016/j.gene.2015.04.032
[9]. Methylation Sequencing. (2023). Illumina; Illumina, Inc. https://doi.org/10.1016/j.gene.2015. 04.032
[10]. Zuo, T., Tycko, B., Liu, T.-M., Lin, H.-J., & Huang, T. (2009). Methods in DNA methylation profiling. Epigenomics, 1(2). https://doi.org/10.2217/epi.09.31
[11]. Frommer, M., McDonald, L., Millar, D., Collist, C., Watt, F., Grigg, G., Molloy, P., & Paul, C. (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Genetics, 89, 1827–1831.
[12]. Whole-genome Bisulfite Sequencing. (2023). Eurofins Genomics; Eurofins Genomics Europe Shared Services GmbH. https://doi.org/10.1101/gr.266551.120
[13]. Vaisvila, R., Ponnaluri, V. K. C., Sun, Z., Langhorst, B., Saleh, L., Guan, S., Dai, Nan, Campbell, Matthew, Sexton, Brittany, Marks, K., Samaranayake, M., Samuelson, J., Church, H., Tamanaha, E., Corrêa Jr., Ivan, Pradhan, S., Dimalanta, Eileen, Evans Jr., T., Williams, L., & Davis, T. (2021). Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res., 31, 1280–1289. https://doi.org/10.1101/gr.266551.120