







1. Introduction
Cells are the basic bricks of living organisms. Within each cell, many distinct intracellular compartments exist to keep the cells working functionally. The main organelles consist of cytosol, endoplasmic reticulum, Golgi apparatus, nucleus, mitochondrion, endosome, lysosome, and peroxisome. The bilayer membrane is the fundamental structure of these organelles and cells themselves and is also a necessary condition for cell survival. Among all these compartments, mitochondria are the vital organelles that power the cells through ATP production. In eukaryotic cells, most of the ATP that drives life processes is produced by mitochondria through oxidative phosphorylation, which certainly has a bilayer membrane structure. They regulate energy homeostasis to maintain cell function and the overall health of the organism. While the mitochondria’ roles in cellular metabolism and energy homeostasis were carefully studied, emerging studies in the past two decades have elucidated the novel functions in many other cellular processes, including cell death, signaling, and cell proliferation. These cellular processes contribute to many complex biological conditions, such as aging.
Mitochondria are multifunctional organelles essential for energy production, metabolism, calcium regulation, and other cellular processes. Because of their central role in energy production, they are often referred to as ‘ cell power plants’. They play a role in ATP production, metabolic regulation, reactive oxygen species ( ROS ) production, calcium regulation, apoptosis ( programmed cell death ), energy homeostasis, and thermogenesis.
Mitochondria dysfunction refers to a condition where the mitochondria, which are the tiny structures within cells responsible for generating energy in the form of adenosine triphosphate (ATP), are not functioning properly. The ability of mitochondria to generate ATP appropriately based on their energy needs. Mitochondrial dysfunction can have many adverse consequences, such as energy depletion, muscle weakness, neurological symptoms, and organ dysfunction. Mitochondrial dysfunction may occur due to genetic mutations, oxidative stress, environmental factors, aging, etc.
Cellular senescence has traditionally been considered a stress response that protects organisms by limiting the proliferation of damaged and unwanted cells. It is also considered a developmental force that is subsequently adapted to protect cells against damage [1]. This process including replication aging and stress-induced premature aging, represents a state of permanent cell growth arrest [2, 3]. Cellular senescence is believed to affect the aging process, but its specific process and mechanism are not yet fully understood.
Aging is defined as the time-related deterioration of the physiological functions necessary for survival and fertility. Aging can be called a chronic disease engraved in genes. Aging is a complex biological process characterized by the gradual decline of various physiological functions, which can affect multiple aspects of personal health. It is related to a series of general age-related changes, including the accumulation of cell damage, changes in gene expression, and a decrease in tissue regeneration. However, the process of aging is extremely complex and changeable. Millions of substances interact to form this complex aging steady state, and mitochondria are part of it.
Mitochondria play an essential role in aging. As the powerhouse of the cell, aged mitochondria lost the capacity to maintain general cellular energy homeostasis, leading to reduced ATP production, increased oxidative stress, alterations in cellular metabolism, and impaired cellular signaling. Senescence also drives mitochondrial dysfunction, causing dysfunction such as genomic instability, impairment of mechanisms involved in mitochondrial mass homeostasis, dysregulated nutrient sensing pathways, imbalanced NAD+/NADH ratio, and calcium overload [4].Mitochondria maintain direct contact with the endoplasmic reticulum [5]and nucleus [6]. These physical contacts are essential in regulating the structure and function of these organelles. Such regulation is disrupted with age. Lysosomes and other organelles also play a vital role in mitochondrial aging [7]. Studies have shown that interventions targeting mitochondrial-related pathways slow or reverse the age-related decline [8]. Understanding the role of mitochondria in aging is particularly important given the increasing aging of the population and the associated burden on the healthcare system.
In this article, we will elaborate on the profound relationship between mitochondria and aging through the following aspects: the structure and function of mitochondria, mitochondrial dysfunction during aging, the relationship between mitochondria and other organelles, and targeted therapy for mitochondria. We will discuss some ideas for future research and development according to the current situation.
2. Mitochondrial Structure and Function
Mitochondria are specialized organelles found in most eukaryotic cells responsible for generating ATP, the primary energy source for cellular activities. Mitochondria have a double membrane system, with the outer membrane being smooth and the inner membrane containing numerous folds called cristæ. The intermembrane space between the two membranes and the matrix inside the inner membrane play essential roles in mitochondrial function.
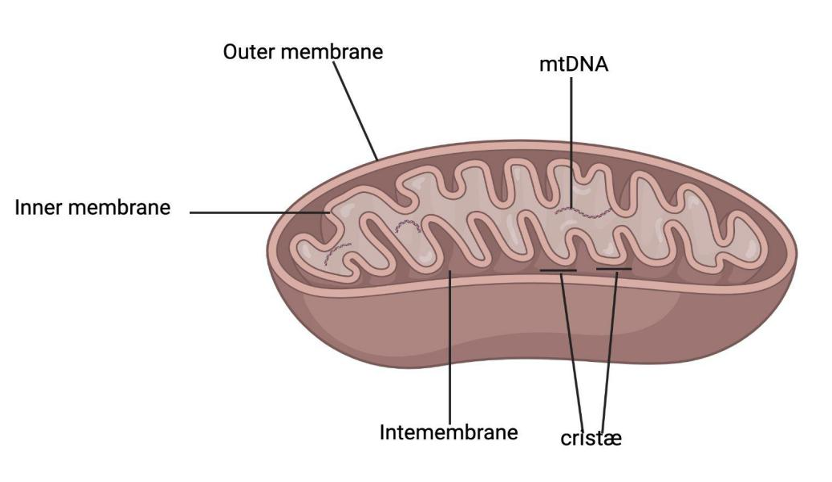
Figure 1. The structure of mitochondria
The outer membrane of mitochondria is smooth and porous, allowing ions, small molecules, and some proteins to pass through. The inner membrane of mitochondria is highly folded, forming a structure called Cristæ. These folds significantly increase the surface area of the inner membrane, thereby achieving more effective ATP production. The inner membrane is impermeable to most ions and molecules, resulting in the proton gradient used in ATP synthesis. Intermembrane is the space between the outer and inner membranes of mitochondria. It contains enzymes involved in various metabolic reactions. Cristæ is a plethora of inner membrane folds containing proteins involved in electron transport chains and ATP synthesis. Most ATP is generated here. Mitochondria have their own DNA molecules, which encode essential proteins related to their electron transport chains and replication mechanisms.
Mitochondria produce ATP through oxidative phosphorylation, which involves the transfer of electrons from electron donors (such as NADH) to electron acceptors in the electron transport chain (ETC) located in the inner mitochondrial membrane. This transfer of electrons generates a proton gradient across the inner mitochondrial membrane, which is used to power the ATP synthase enzyme to produce ATP from ADP and inorganic phosphate. The final electron acceptor in the ETC is oxygen, which combines with protons to create water [9]. This process is highly efficient and generates the vast majority of ATP used by cells.
However, reactive oxygen species (ROS) can also be generated during oxidative phosphorylation as a byproduct of electron transfer reactions in the ETC. Specifically, electrons can leak out of the ETC before reaching the final acceptor (oxygen) and react with molecular oxygen to form superoxide, a highly reactive ROS. Superoxide can then undergo further reactions to form other ROS, such as hydrogen peroxide and hydroxyl radicals. Mitochondrial ROS-mediated signaling pathways can participate in various basic and adaptive physiological responses that control homeostasis [10]. Complex I and III are usually considered the main sites for Mitochondrial ROS (mtROS) production, but recent studies have shown that at least 10 other mitochondrial enzymes also contribute to this process, including complex II. The different sites that produce mtROS have different signaling effects, and the main production sites may change under different physiological conditions [11]. Researchers argue for the key correlation of mtROS signal transients as important contributors to longevity, as well as mediators of other signaling responses that promote health span and longevity [12].
Mitochondria generate the majority of ATP through oxidative phosphorylation, essential for energy-consuming cellular processes. Mitochondria also play important roles in cellular signal transduction. Mitochondrial signal transduction happens in a three-step process, mitochondria (1) sense and respond to both endogenous and environmental inputs, like metabolism and hormone inputs, through morphological and functional remodeling; (2) integrate information through dynamic, network-based physical interactions and diffusion mechanisms; and (3) produce output signals that tune the functions of other organelles and systemically regulate physiology [13], for example, punctual, localized, and pulsatile redox-based communication between mitochondria and the ER can propagate signals from single mitochondria to the ER and other mitochondria [14].
In summary, mitochondria are the cellular energy source in most eukaryotic cells, and play a crucial role in the production of ATP, the main energy source, through oxidative phosphorylation. They have a unique double-membrane structure with internal folds called ridges. This energy generation involves the transfer of electrons in the electron transport chain, resulting in a proton gradient that provides energy for ATP synthesis. However, this process also produces reactive oxygen species ( ROS ), affecting cell signal transduction and physiological responses. Mitochondria are also involved in cell signal transduction, sensing, and response to metabolic inputs, and produce signals that regulate various cell functions and physiology through dynamic interactions with other organelles.
3. Mitochondrial dysfunction, cell senescence, and aging
Mitochondrial dysfunction refers to damage to the structure or function of mitochondria. This dysfunction may be caused by factors such as oxidative stress, DNA mutations, impaired mitochondrial quality control mechanisms, mitochondrial protein, and lipid damage.
Mitochondria are key organelles for aerobic respiration and cellular energy metabolism, and some mitochondrial structures and functions related to energy production are closely related to aging and senescence. The mitochondrial membrane potential (MMP) is a critical process of mitochondrial function, and the two are closely interrelated. The proton gradient generated by MMP serves as the driving force for ATP synthesis, and healthy MMP ensures effective electron transport, and ATP production, and prevents excessive production of reactive oxygen species (ROS). The instantaneous reduction of MMP puts the electron transport chain (ETC) in a more oxidized state and can reduce the production of ROS, but the continuous reduction of MMP may indicate ETC dysfunction. Due to the oxidation of the reduced form of NADH to NAD+ by complex I in ETC, a decrease in respiratory capacity may contribute to a decrease in NAD+/NADH balance during aging and cellular senescence [15]. A decrease in MMP in a stable state may indicate impaired electron transfer chain function or increased proton leakage. During mitochondrial dysfunction, low MMP is usually associated with increased ROS production [16]. Cell senescence may be accompanied by changes in mitochondrial oxidative phosphorylation (OXPHOS). ROS links mitochondrial dysfunction with telomere dependence and pro-inflammatory aging. Damaged mitochondria with inefficient OXPHOS produce excessive ROS. Increased oxidative stress can lead to DNA damage, such as oxidative bases, single-strand breaks, double-strand breaks, and telomere shortening [17]. The production of cellular ATP in young primary human brain microvascular endothelial cells (HBMEC) mainly relies on glycolysis, and glutamine is the preferred fuel for mitochondrial OXPHOS. In contrast, HBMEC before aging contributes equally to the glycolysis and ATP production rate of OXPHOS and also utilizes glutamine, glucose, and fatty acids as mitochondrial fuels [18]. There are widely varying results regarding the importance of individual ETC complexes in respiratory loss. For example, complex I am usually more sensitive to age-related functional loss. As the largest respiratory chain complex, the function of complex I and the production of ROS depend on assembly fidelity, which decreases with age, while the knockdown of individual complex I assembly factors is sufficient to induce cell senescence [19]. The respiratory chain complex forms a super complex, and its stability seems to decrease with age [20].
Mitochondria are very dynamic organelles, and maintain the continuous remodeling of mitochondrial structure, allowing morphological transformation from a single structure to a complex tubular network through fusion and fission [21]. Recent studies have shown that mitochondria interact extensively with the endoplasmic reticulum and the nucleus [22] [23]. Changes in these interactions may lead to neurodegeneration. Mitochondria have their own DNA (mtDNA) and are easily damaged. Accumulated mitochondrial DNA mutations or deletions can lead to mitochondrial dysfunction and senescence, and mtDNA mutations accumulate with age in tissues after mitosis [24]. Studies on mtDNA polymerase proofreading activity defects have demonstrated the mechanism of mtDNA damage and aging. Research has shown that at high densities, mtDNA mutations can lead to physiological-related mitochondrial dysfunction and premature aging [25]. The mitochondrial genome is important in maintaining a functionally competent organelle. With age, there is a gradual decrease in mitochondrial function, leading to reduced ATP production. This decline is often accompanied by an increase in oxidative stress, which can damage mitochondrial DNA (mtDNA) and proteins, further impairing mitochondrial function [26].
Mitochondrial quality can be regulated through the process of mitochondrial autophagy, which involves the selective degradation of damaged or dysfunctional mitochondria. In senescent cells, mitochondrial autophagy may be damaged, leading to the accumulation of damaged mitochondria and changes in mitochondrial quality. This means that the efficiency of mitochondria in producing ATP through oxidative phosphorylation is low, leading to a decrease in energy production. Research has shown that reducing mitochondrial content in the body can prevent the aging of the liver in aging mice [27]. In vitro experiments, mitochondrial autophagy decreases during aging, leading to the accumulation of dysfunctional mitochondria. High expression of some proteins, such as S-nitroso glutathione reductase (GSNOR), may promote cell longevity by maintaining mitochondrial dynamics and preventing mitochondrial autophagy from being damaged [28].
Senescence drives mitochondrial dysfunction, while Mitochondrial dysfunction governs the senescent phenotype. Some material and structural changes that occur during aging and cellular senescence may lead to mitochondrial dysfunction. Several metabolic regulatory factors of nutritional sensing mechanisms, such as insulin/IGF-1, mTOR, AMPK, and sirtuins, are all associated with aging. Excessive cellular nutrients activate the insulin/IGF-1 and mTOR pathways, leading to the induction of synthetic metabolic processes and inhibition of autophagy [29]. Sirtuins are NAD+ dependent deacetylases and metabolic sensors that play important roles in stress and cellular metabolism. Sirtuins regulate the biogenesis of mitochondria and regulate their composition and function. In mammals, the three members of the SIRT family, SIRT3, SIRT4, and SIRT5, are located in mitochondria and regulate mitochondrial metabolism [30]. The decrease in NAD+ regulated by CD38 and mitochondrial dysfunction in senescence is at least partially mediated by SIRT3 [31].NAD+ acts as a cofactor in many oxygen reduction pathways and as a substrate in some redox reactions. Both ATP production and MMP maintenance require NAD+. Recent studies have reported the role of NAD+ consuming enzyme CD38 in the decline of NAD+ during aging [32]. The expression and activity of CD38 are induced during the aging process in chronological order, and this increase in CD38+ cells is partially mediated by the SASP of senescent cells, indicating a strong connection between cell aging and a decrease in NAD+ during the aging process [33]. Mitochondrial Ca2+ influx occurs through a voltage-dependent anion channel (VDAC) present in the OMM, and then Ca2+ enters the mitochondrial matrix through the mitochondrial calcium single transporter (MCU) located in the IMM. The intracellular Ca2+ buffer is controlled by the interaction of 1,4,5-triphosphate inositol receptor (IP3R), Grp75, and VDAC [34]. The increase in cytoplasmic Ca2+ concentration leads to rapid uptake of Ca2+ by mitochondria to prevent Ca2+ overload in the cytoplasm but may lead to mitochondrial Ca2+ overload [35], which leads to increased ROS production and mitochondrial dysfunction, including reduced ATP production [36]. Mitochondrial Ca2+ overload causes mitochondrial metabolic damage during aging and age-related diseases [37]During aging, the activation of IP3R leads to the release of Ca2+ from the endoplasmic reticulum and the accumulation of Ca2+ through MCU channels, leading to mitochondrial Ca2+ overload [38]. The overload of Ca2+ in mitochondria leads to a decrease in membrane potential, an increase in ROS production, and senescence.
Cell senescence and mitochondrial dysfunction are defined as classic features of the aging process. The relationship between them is very complicated. It is now widely believed that cellular senescence is a key driver of aging and many age-related diseases. Cell senescence is the central marker of aging. Telomere damage, epigenetic disorders, DNA damage, and mitochondrial dysfunction are the main drivers of damage during aging. Several of these damage drivers can induce cell senescence. Cell senescence can in turn drive subsequent aging characteristics to cope with damage: stem cell exhaustion and chronic inflammation [39]. Firstly, emerging evidence has pinpointed mitochondria as one of the key modulators in the development of the senescence phenotype, particularly the pro-inflammatory senescence-associated secretory phenotype (SASP) [40]. Secondly, Mitochondrial dysfunction leads to ROS accumulation, leading to oxidative stress and damage to cellular components. Accumulated DNA damage, usually caused by ROS, is a well-known trigger of cellular senescence [41]. Thirdly, maintaining healthy mitochondria through processes such as mitophagy ( selective removal of damaged mitochondria ) is essential to prevent dysfunctional mitochondrial accumulation and dysregulation of mitophagy and mitochondrial quality control mechanisms may lead to mitochondrial dysfunction and may promote cell senescence. During senescence, mitophagy is disrupted, leading to mitochondrial accumulation and Senescence-Associated Mitochondrial Dysfunction (SAMD) [42].
Mitochondrial dysfunction has been widely implicated in the aging process and the development of aging-related diseases. Many studies have provided evidence of mitochondrial dysfunction in aging. Early studies on mitochondria have pointed out age-related changes in muscle mitochondria of insects ( such as flies ), including a decrease in the number of mitochondria, accompanied by an increase in residual mitochondria and more irregular structures, loss of mitochondrial cristae stacking arrays, and destruction of crystal accumulation with the expansion of space within the cristae [43]. Mitochondrial dysfunction is more likely to be a part contributing to aging because aging is more like a macroscopic phenomenon caused by the decline in the function of multiple cells and tissues.
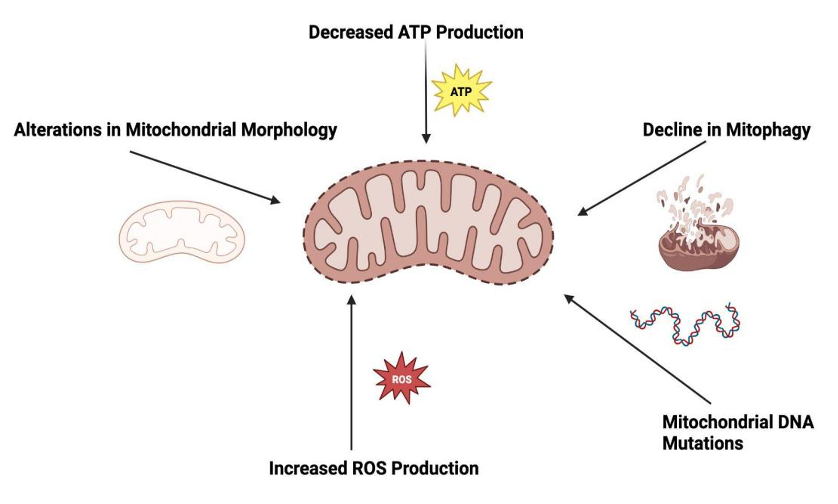
Figure 2.The potential mechanisms of mitochondrial dysfunction during aging
During the aging process, mitochondrial dysfunction is reflected in the following aspects: 1. Reduced ATP production: Aging leads to a decrease in the efficiency of ATP production through oxidative phosphorylation within mitochondria. This decrease helps to lower cellular energy levels, which may lead to fatigue and reduce body performance. 2. Increased ROS production: Mitochondria, generators, and targets of reactive oxygen species (ROS), have lower efficiency in electron transport chains with age. This leads to an increase in ROS production, leading to oxidative damage to mitochondrial components and further impairing their function. 3. Morphological changes: Aging can trigger changes in mitochondrial structure, leading to changes in size and shape. These morphological transfers can affect mitochondrial function and dynamics, including regulating the fusion and fission processes of organelle structures. 4. Decreased mitochondrial autophagy: The efficiency of mitochondrial autophagy (selective removal of damaged mitochondria) decreases with age. Inefficient mitochondrial autophagy leads to the accumulation of dysfunctional mitochondria, exacerbating mitochondrial dysfunction. 5. mtDNA mutation: Mitochondria contain their own DNA (mtDNA) and are susceptible to oxidative damage. Over time, mtDNA accumulates mutations that hinder mitochondrial function. These mutations may be genetic and related to mitochondrial diseases.
In summary, Mitochondrial dysfunction encompasses structural and functional impairments in mitochondria, arising from factors like oxidative stress, DNA mutations, disrupted quality control mechanisms, and damage to mitochondrial proteins and lipids. These crucial organelles play a pivotal role in cellular energy metabolism and aging processes, particularly through their membrane potential (MMP) and electron transport chain (ETC) functions. Altered MMP can indicate ETC dysfunction, contributing to reduced NAD+/NADH balance, increased ROS production, and cellular senescence. Mitochondrial dysfunction also links to oxidative phosphorylation (OXPHOS) changes, DNA damage, and pro-inflammatory aging. Additionally, the dynamics of mitochondrial structure, interactions with the endoplasmic reticulum and nucleus, and mitochondrial DNA mutations all play significant roles in aging-related mitochondrial dysfunction. Mitochondrial autophagy, vital for maintaining mitochondrial quality, can become impaired in senescent cells, leading to the accumulation of damaged mitochondria and decreased energy production. Strategies to address mitochondrial dysfunction may hold promise in mitigating age-related health issues. The intricate relationship between cellular senescence and mitochondrial dysfunction plays a central role in the aging process and age-related diseases. While cellular senescence is recognized as a driving force behind aging and age-related ailments, mitochondria emerge as pivotal modulators of the senescence phenotype, particularly the pro-inflammatory senescence-associated secretory phenotype (SASP). Mitochondrial dysfunction results in the accumulation of reactive oxygen species (ROS), oxidative stress, and damage to cellular components, including DNA damage, which is a known inducer of cellular senescence. Maintaining healthy mitochondria through processes like mitophagy is crucial to prevent dysfunctional mitochondrial buildup, and disturbances in mitophagy and mitochondrial quality control mechanisms can lead to mitochondrial dysfunction and promote cell senescence, a phenomenon referred to as Senescence-Associated Mitochondrial Dysfunction (SAMD). Mitochondrial dysfunction is widely implicated in aging and age-related diseases, with studies revealing age-related changes in mitochondrial structure and function. Ultimately, mitochondrial dysfunction appears to be a contributing factor to the aging process, as aging involves the decline in the function of multiple cells and tissues, with mitochondrial dysfunction playing a significant role.
4. Inter-organelle relationship
Mitochondria are not isolated entities within cells. They interact with various organelles in complex ways to shape cellular function and maintain internal balance. The organelles most closely related to mitochondria are mainly the nucleus and endoplasmic reticulum. Other organelles in eukaryotic cells, such as the Golgi apparatus, lysosomes, and peroxisomes, also have some Inter-organelle relationship with mitochondria.
The close contact between the endoplasmic reticulum (ER) and mitochondria, often referred to as the “mitochondrial-associated membrane” or MAM is a specialized subdomain where the membranes of these two organelles come into proximity but do not fuse. The normal function of mitochondria requires the participation of other organelles, and the mitochondria endoplasmic reticulum contact site is closely related to lipid biosynthesis, Ca2+ signaling, mitochondrial fission and fusion, and other processes [44]. The mass spectrometric analysis enabled the proteins identified in MAM to be characterized and divided into three groups: (1) proteins localized only in MAM (‘MAM resident proteins’); (2) the protein localizes to mam, but is present in other regions of (‘MAM enriched proteins’); (3) temporarily present in MAM (‘MAM associated protein’) [45]. Mitochondria-endoplasmic reticulum contacts (MERCs) are a subset of MAMs and are often used when discussing the interaction and functional coordination between mitochondria and the endoplasmic reticulum specifically. Membrane contact sites have emerged in the past decade and also become key participants in the integration, regulation, and transmission of many intracellular signals, playing a crucial role in various pathological and physiological environments. Numerous studies have pointed out the role of mitochondrial endoplasmic reticulum contact (MERC) in regulating aging. Nevertheless, the driving cellular mechanism behind this effect is still unclear. Recent evidence suggests that MERC regulates cell senescence, a permanent proliferative arrest state associated with pro-inflammatory secretion, which may mediate the impact of MERC on aging [46]. The distance between ER and mitochondria can be measured using advanced methods, based on the width of the cracks separating the mitochondrial outer membrane (OMM) from ER, and MERC can be subdivided into tight (~10 nm) and loose (~25-40 nm) structures [47]. The contact between mitochondria and endoplasmic reticulum is essential for mitochondrial division and fusion. The endoplasmic reticulum (ER) does not directly participate in mitochondrial division and fusion processes, but it can influence these processes indirectly through its interactions with mitochondria at mitochondria-associated membrane (MAM) contact sites. Mitochondrial fission(division) is the process of dividing a single mitochondrial into two or smaller mitochondria. This process is regulated by proteins such as Dynamin-related protein 1 (Drp1), which contracts and cleaves mitochondria [48].Mfn1 and Mfn2 are proteins on the mitochondrial inner membrane (IMM) and are associated with mitochondrial fusion. Studies have shown that the levels of Mfn1 and Mfn2 increase in aging skeletal muscles, indicating upregulation of fusion [49]. Autophagy is a tightly regulated intracellular large-scale degradation/recycling system that plays an important role in cellular homeostasis. Recent studies have shown that autophagosomes form at the endoplasmic reticulum mitochondria contact sites in mammalian cells. [5] Basic autophagy levels are essential for physiological quality control, but autophagy’s damage and decreased efficacy are related to many human pathologies and aging [50].
Another organelle that interacts with mitochondria is the nucleus. Mitochondria have their own DNA (mtDNA), which encodes the set of protein protons necessary for mitochondrial function. However, most mitochondrial proteins are encoded by nuclear DNA. The first step in mitochondrial nuclear interaction is the transcription and translation of mitochondrial proteins encoded by nDNA in the cytoplasm. These proteins are then input into mitochondria to support their biogenesis, maintenance, and function. Proteins synthesized in the cytoplasm are called mitochondrial precursor proteins, which are transported to mitochondria through specialized input pathways. The Translocase of the Outer Membrane (TOM) complex is responsible for the initial recognition and transportation of these precursor proteins through the mitochondrial outer membrane. Then, the protein is directed to the mitochondrial intima and matrix through additional transposase complexes, such as Translocase of the Inner Membrane (TIM) complexes [51]. Mitochondria have their own DNA(mt DNA), but many of the substances required for mitochondrial synthesis still come from the nucleus, and there are different feedback regulations between them [52]. The function of mitochondria is strictly regulated by nuclear activity. It requires extensive communication between these organelles, Cnm1 (contact nucleus mitochondria 1)is a protein on the nuclear membrane that mediates contact by interacting with Tom70 on mitochondria [53].
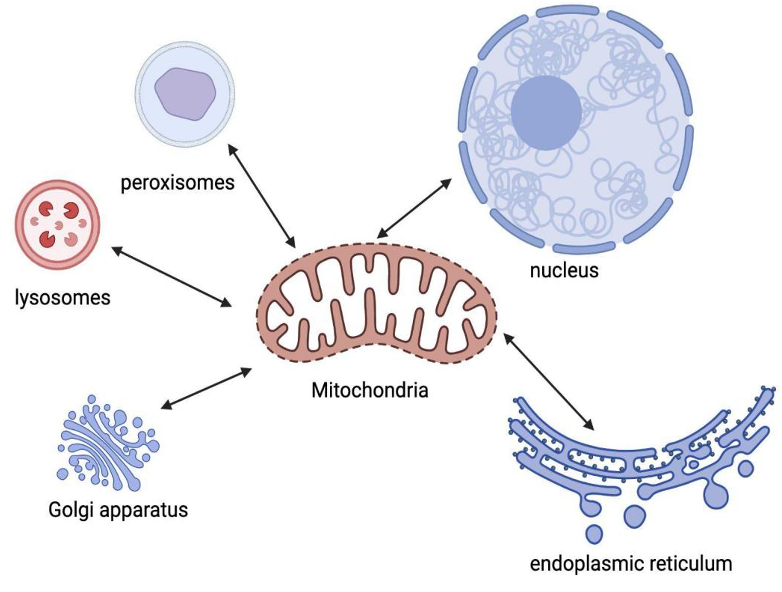
Figure 3. Inter-organelle relationship of mitochondria and other organelles.
Mitochondria are not isolated entities in cells, and the organelles most closely related to mitochondria are mainly the nucleus and endoplasmic reticulum. The Golgi apparatus, lysosomes, and peroxisomes also have some interactions with mitochondria. Mitochondria interact closely with endoplasmic reticulum ( complex membrane network in cells ). This interaction is known as mitochondrial-associated endoplasmic reticulum ( MAM ). It plays a vital role in lipid metabolism, calcium regulation, and the exchange of various molecules between two organelles. The interaction between mitochondria and the nucleus is mainly reflected in the synthesis of proteins and enzymes. Mitochondria contain their own DNA ( mitochondrial DNA or mtDNA ), which can be replicated independently of the cell cycle. However, most of their proteins and enzymes depend on the nucleus. The nucleus controls the synthesis and import of these proteins into mitochondria.
Cells are a complex whole, and there are countless signaling pathways and material compositions between mitochondria and other organelles. The steady-state shift of these pathways and substances plays an important role in cellular aging and overall aging.
Inter-organelle interactions profoundly impact mitochondrial function and aging. ER-mitochondria crosstalk influences calcium dynamics, affecting mitochondrial energy production and ROS generation. MCU(mitochondrial calcium uniporter) is a unidirectional transport protein for the mitochondrial uptake of Ca2+. The increased ER-mitochondria Ca2+ transfer was accompanied by the upregulation of the mitochondrial calcium uniporter (MCU) [54]. The function of this channel protein is closely related to the endoplasmic reticulum. Aging is often accompanied by progressive organ failure. Overexpression of MCU saves muscle and reverses cardiac failure by reducing Ca2+ leakage in the endoplasmic reticulum [55]. Sometimes mitochondria and endoplasmic reticulum form contact point, which are called mitochondria-ER contacts(MERCs). Research has shown that the microstructure of MERCs themselves, especially their thickness, is a key factor in regulating Ca2+ transport efficiency [56]. The knockout of MCU and inositol 1,4,5-triphosphate receptor type 2 (ITPR2) is involved in the accumulation of calcium in mitochondria, leading to aging escape, showing that mitochondrial calcium accumulation plays a role in aging induction [57]. The balance of Ca2+ is crucial for ROS, as they are also closely related to aging and age-related diseases [58].
Basic autophagy levels are essential for physiological quality control, but autophagy’s damage and decreased efficacy are related to many human pathologies and imaginations [59]. The lysosomal mitochondrial axis plays a crucial role in mitochondrial quality control through mitochondrial autophagy (selective degradation of damaged mitochondria). The imbalance of this process can lead to the accumulation of dysfunctional mitochondria, exacerbating aging-related decline [60]. Mitochondrial DNA (mtDNA) encodes 13 proteins crucial for electron transport, as well as genes encoding 12S and 16S rRNAs and 22 transfer RNAs. Mutations of mtDNA accumulate with age in tissues after mitosis [61], and there are crosstalks between the repair of nuclear and mtDNA [62]. The nuclear-mitochondrial interaction regulates mitochondrial biogenesis and the expression of nuclear-encoded mitochondrial genes. The damage to mitochondrial function caused by mtDNA and nuclear DNA mutations may lead to imbalanced cellular energy homeostasis, increased vulnerability to oxidative stress, and accelerated cellular aging and aging [63].
5. Mitochondrially targeted intervention
The substances produced by mitochondria or the functions involved in regulation are important for both cellular senescence and aging, therefore, it is necessary to explore targeted interventions for mitochondria. Mitochondria are organelles responsible for producing energy in the form of adenosine triphosphate (ATP). However, as we age, mitochondria often accumulate damage, leading to dysfunction.
The current strategies to address mitochondrial dysfunction during aging include lifestyle modifications, pharmaceutical interventions, antioxidants, and mitochondrial biogenesis. In early studies, it was found that limiting calorie intake can prolong the lifespan of experimental rats [64]. Calorie-restricted feeding can slow down the rate of oxidative damage, as the mitochondria of these animals produce superoxide at a lower rate compared to the mitochondria of the control animals [65]. Exercise strategies can also be used to improve/maintain mitochondrial health during the aging process [66]. Some drugs, such as coenzyme Q10, have been explored for their potential to support mitochondrial health [67]. Antioxidants (such as vitamin C) and other compounds are used to counteract oxidative stress that damages mitochondria [68], the concentration of these oxidants in mitochondria is relatively low, so there is almost no therapeutic effect targeting mitochondria. As for mitochondrial biogenesis, activating pathways like PGC-1α can enhance the production of new, healthy mitochondria [69].
The structural and functional changes of mitochondria are related to many age-related diseases, and abnormal mitochondrial dysfunction may lead to abnormal cell proliferation and survival, which contributes to tumor formation. Neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease are also associated with mitochondrial dysfunction. Mitochondria’s role in providing energy and protecting nerve cells from oxidative stress is critical. When mitochondria don’t function properly, it can cause nerve cell damage and death, advancing neurodegenerative diseases. In terms of cardiovascular disease, cardiac muscle cells require a large amount of energy to maintain the pumping function of the heart. Impaired mitochondrial function may lead to a decrease in cardiac muscle cell function, increasing the risk of cardiovascular disease, such as myocardial infarction and heart failure. Therefore, maintaining healthy mitochondrial function may be crucial for preventing or delaying the aging process associated with cancer, neurodegenerative diseases, and cardiovascular diseases. This includes adopting healthy lifestyle choices such as a balanced diet and moderate exercise, as well as possible drug interventions to support mitochondrial function.
Improving mitochondrial function can have various benefits such as improving energy production, reducing oxidative stress, and protecting neurons from degeneration. However, individual specificity, drug safety and long-term impact, and regulatory approval may be challenges that hinder the further development of mitochondrial-targeted drugs and need to be carefully addressed in ongoing research and development work.
6. Conclusion
This article delves into the complex relationship between mitochondria and the aging process. Mitochondria, commonly known as cellular power banks, are the foundation of energy production and have a significant impact on various cellular functions. Mitochondrial dysfunction may be caused by factors such as oxidative stress and gene mutations and is a core factor in age-related health problems. Although cell aging is related to the cessation of cell growth, its mechanism is still not fully understood.
Aging is a complex process characterized by a gradual decline in the basic functions required for survival and reproduction. Mitochondria undergo structural and functional changes with age, and they interact with various cellular structures, which evolve with age. With the aging population and the increasing demand for healthcare, this understanding has become increasingly important.
Mitochondrial dysfunction, characterized by injury or impairment, is a key factor in the aging process. It affects factors such as mitochondrial membrane potential (MMP), energy production, and reactive oxygen species (ROS) management. Maintaining mitochondrial quality control is crucial, and deviations in this process may lead to energy deficiency, leading to the aging process. In addition, mitochondrial dysfunction is closely related to cellular aging, which is a key aspect of aging.
The complex interplay between mitochondrial dysfunction, aging, and organ interactions has a profound impact on cellular senescence and age-related diseases. Strategies to address mitochondrial dysfunction during aging include lifestyle changes, drug interventions, antioxidants, and promoting healthy mitochondrial growth. These interventions aim to enhance mitochondrial function, reduce oxidative stress, and prevent health issues related to aging. However, addressing individual variability and ensuring drug safety are the most important considerations when developing mitochondrial-targeted therapies for healthy aging.
References
[1]. Da Silva-Álvarez S, Picallos-Rabina P, Antelo-Iglesias L, et al. The development of cell senescence. Experimental Gerontology 2019; 128: 110742.
[2]. Salama R, Sadaie M, Hoare M, et al. Cellular senescence and its effector programs. Genes Dev 2014; 28: 99–114.
[3]. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8: 729–740.
[4]. Miwa S, Kashyap S, Chini E, et al. Mitochondrial dysfunction in cell senescence and aging. Journal of Clinical Investigation 2022; 132: e158447.
[5]. Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013; 495: 389–393.
[6]. Walker BR, Moraes CT. Nuclear-Mitochondrial Interactions. Biomolecules 2022; 12: 427.
[7]. Hughes CE, Coody TK, Jeong M-Y, et al. Cysteine Toxicity Drives Age-Related Mitochondrial Decline by Altering Iron Homeostasis. Cell 2020; 180: 296-310.e18.
[8]. Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Molecular Cell 2016; 61: 654–666.
[9]. Bonora M, Patergnani S, Rimessi A, et al. ATP synthesis and storage. Purinergic Signalling 2012; 8: 343–357.
[10]. Shadel GS, Horvath TL. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015; 163: 560–569.
[11]. Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, et al. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biology 2013; 1: 304–312.
[12]. Schmeisser S, Priebe S, Groth M, et al. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Molecular Metabolism 2013; 2: 92–102.
[13]. Picard M, Shirihai OS. Mitochondrial signal transduction. Cell Metabolism 2022; 34: 1620–1653.
[14]. Booth DM, Várnai P, Joseph SK, et al. Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Molecular Cell 2021; 81: 3866-3876.e2.
[15]. Nacarelli T, Lau L, Fukumoto T, et al. NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol 2019; 21: 397–407.
[16]. Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Molecular Cell 2016; 61: 654–666.
[17]. Von Zglinicki T. Oxidative stress shortens telomeres. Trends in Biochemical Sciences 2002; 27: 339–344.
[18]. Sakamuri SSVP, Sure VN, Kolli L, et al. Glycolytic and Oxidative Phosphorylation Defects Precede the Development of Senescence in Primary Human Brain Microvascular Endothelial Cells. GeroScience 2022; 44: 1975–1994.
[19]. Miwa S, Jow H, Baty K, et al. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat Commun 2014; 5: 3837.
[20]. Ramírez-Camacho I, Flores-Herrera O, Zazueta C. The relevance of the supramolecular arrangements of the respiratory chain complexes in human diseases and aging. Mitochondrion 2019; 47: 266–272.
[21]. Chen H, Chan DC. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Human Molecular Genetics 2009; 18: R169–R176.
[22]. Mitochondria form contact sites with the nucleus to couple prosurvival retrograde response. SCIENCE ADVANCES 2020; 16.
[23]. Wilson EL, Metzakopian E. ER-mitochondria contact sites in neurodegeneration: genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ 2021; 28: 1804–1821.
[24]. Simon DK, Lin MT, Zheng L, et al. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson’s disease. Neurobiology of Aging 2004; 25: 71–81.
[25]. Trifunovic A, Hansson A, Wredenberg A, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA 2005; 102: 17993–17998.
[26]. Pinto M, Moraes CT. Mechanisms linking mtDNA damage and aging. Free Radical Biology and Medicine 2015; 85: 250–258.
[27]. Correia‐Melo C, Marques FD, Anderson R, et al. Mitochondria are required for pro‐ageing features of the senescent phenotype. The EMBO Journal 2016; 35: 724–742.
[28]. Rizza S, Cardaci S, Montagna C, et al. S -nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc Natl Acad Sci USA; 115. Epub ahead of print 10 April 2018. DOI: 10.1073/pnas.1722452115.
[29]. Johnson SC. Nutrient Sensing, Signaling and Ageing: The Role of IGF-1 and mTOR in Ageing and Age-Related Disease. In: Harris JR, Korolchuk VI (eds) Biochemistry and Cell Biology of Ageing: Part I Biomedical Science. Singapore: Springer Singapore, pp. 49–97.
[30]. Yang W, Nagasawa K, Münch C, et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 2016; 167: 985-1000.e21.
[31]. Camacho-Pereira J, Tarragó MG, Chini CCS, et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metabolism 2016; 23: 1127–1139.
[32]. Chini CCS, Peclat TR, Warner GM, et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nat Metab 2020; 2: 1284–1304.
[33]. Covarrubias AJ, Kale A, Perrone R, et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat Metab 2020; 2: 1265–1283.
[34]. Xu H, Guan N, Ren Y-L, et al. IP3R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol 2018; 19: 140.
[35]. Bravo‐Sagua R, Parra V, López‐Crisosto C, et al. Calcium Transport and Signaling in Mitochondria. In: Terjung R (ed) Comprehensive Physiology. Wiley, pp. 623–634.
[36]. Brookes PS, Yoon Y, Robotham JL, et al. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American Journal of Physiology-Cell Physiology 2004; 287: C817–C833.
[37]. Müller M, Ahumada-Castro U, Sanhueza M, et al. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated With Aging. Front Neurosci 2018; 12: 470.
[38]. Wiel C, Lallet-Daher H, Gitenay D, et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun 2014; 5: 3792.
[39]. McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. Journal of Cell Biology 2018; 217: 65–77.
[40]. Chapman J, Fielder E, Passos JF. Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Letters 2019; 593: 1566–1579.
[41]. Davalli P, Mitic T, Caporali A, et al. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxidative Medicine and Cellular Longevity 2016; 2016: 1–18.
[42]. Korolchuk VI, Miwa S, Carroll B, et al. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017; 21: 7–13.
[43]. Miquel J, Economos AC, Fleming J, et al. Mitochondrial role in cell aging. Experimental Gerontology 1980; 15: 575–591.
[44]. Rowland AA, Voeltz GK. Endoplasmic reticulum–mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 2012; 13: 607–615.
[45]. Poston CN, Krishnan SC, Bazemore-Walker CR. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). Journal of Proteomics 2013; 79: 219–230.
[46]. Ziegler DV, Martin N, Bernard D. Cellular senescence links mitochondria-ER contacts and aging. Commun Biol 2021; 4: 1323.
[47]. Cieri D, Vicario M, Giacomello M, et al. SPLICS: a split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ 2018; 25: 1131–1145.
[48]. Kalia R, Wang RY-R, Yusuf A, et al. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018; 558: 401–405.
[49]. Joseph A-M, Adhihetty PJ, Wawrzyniak NR, et al. Dysregulation of Mitochondrial Quality Control Processes Contribute to Sarcopenia in a Mouse Model of Premature Aging. PLoS ONE 2013; 8: e69327.
[50]. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and Aging. Cell 2011; 146: 682–695.
[51]. Rassow J, Dekker PJT, Van Wilpe S, et al. The preprotein translocase of the mitochondrial inner membrane: function and evolution. Journal of Molecular Biology 1999; 286: 105–120.
[52]. Poyton RO, McEwen JE. CROSSTALK BETWEEN NUCLEAR AND MITOCHONDRIAL GENOMES. Annu Rev Biochem 1996; 65: 563–607.
[53]. Eisenberg-Bord M, Zung N, Collado J, et al. Cnm1 mediates nucleus–mitochondria contact site formation in response to phospholipid levels. Journal of Cell Biology 2021; 220: e202104100.
[54]. Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. The Journal of Physiology 2014; 592: 829–839.
[55]. Liu T, Yang N, Sidor A, et al. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca 2+ Leak. Circ Res 2021; 128: 1191–1204.
[56]. Giacomello M, Pellegrini L. The coming of age of the mitochondria–ER contact: a matter of thickness. Cell Death Differ 2016; 23: 1417–1427.
[57]. Wiel C, Lallet-Daher H, Gitenay D, et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun 2014; 5: 3792.
[58]. Madreiter-Sokolowski CT, Thomas C, Ristow M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox Biology 2020; 36: 101678.
[59]. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and Aging. Cell 2011; 146: 682–695.
[60]. Park JT, Lee Y-S, Cho KA, et al. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Research Reviews 2018; 47: 176–182.
[61]. Simon DK, Lin MT, Zheng L, et al. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson’s disease. Neurobiology of Aging 2004; 25: 71–81.
[62]. Saki M, Prakash A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radical Biology and Medicine 2017; 107: 216–227.
[63]. Tranah GJ. Mitochondrial–nuclear epistasis: Implications for human aging and longevity. Ageing Research Reviews 2011; 10: 238–252.
[64]. McCay CM, Crowell MF, Maynard LA. The Effect of Retarded Growth Upon the Length of Life Span and Upon the Ultimate Body Size. The Journal of Nutrition 1935; 10: 63–79.
[65]. Merry BJ. Oxidative stress and mitochondrial function with aging - the effects of calorie restriction. Aging Cell 2004; 3: 7–12.
[66]. Kim Y, Triolo M, Hood DA. Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle. Oxidative Medicine and Cellular Longevity 2017; 2017: 1–16.
[67]. Hidalgo-Gutiérrez A, González-García P, Díaz-Casado ME, et al. Metabolic Targets of Coenzyme Q10 in Mitochondria. Antioxidants 2021; 10: 520.
[68]. KC S, Càrcamo JM, Golde DW. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Gluti) and confers mitochondrial protection against oxidative injury. FASEB j 2005; 19: 1657–1667.
[69]. Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. The American Journal of Clinical Nutrition 2011; 93: 884S-890S.
Cite this article
Chen,M. (2024). Mitochondria in aging: A review of structure, function, and interorganelle relationships. Theoretical and Natural Science,29,69-81.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Da Silva-Álvarez S, Picallos-Rabina P, Antelo-Iglesias L, et al. The development of cell senescence. Experimental Gerontology 2019; 128: 110742.
[2]. Salama R, Sadaie M, Hoare M, et al. Cellular senescence and its effector programs. Genes Dev 2014; 28: 99–114.
[3]. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8: 729–740.
[4]. Miwa S, Kashyap S, Chini E, et al. Mitochondrial dysfunction in cell senescence and aging. Journal of Clinical Investigation 2022; 132: e158447.
[5]. Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013; 495: 389–393.
[6]. Walker BR, Moraes CT. Nuclear-Mitochondrial Interactions. Biomolecules 2022; 12: 427.
[7]. Hughes CE, Coody TK, Jeong M-Y, et al. Cysteine Toxicity Drives Age-Related Mitochondrial Decline by Altering Iron Homeostasis. Cell 2020; 180: 296-310.e18.
[8]. Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Molecular Cell 2016; 61: 654–666.
[9]. Bonora M, Patergnani S, Rimessi A, et al. ATP synthesis and storage. Purinergic Signalling 2012; 8: 343–357.
[10]. Shadel GS, Horvath TL. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015; 163: 560–569.
[11]. Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, et al. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biology 2013; 1: 304–312.
[12]. Schmeisser S, Priebe S, Groth M, et al. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Molecular Metabolism 2013; 2: 92–102.
[13]. Picard M, Shirihai OS. Mitochondrial signal transduction. Cell Metabolism 2022; 34: 1620–1653.
[14]. Booth DM, Várnai P, Joseph SK, et al. Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Molecular Cell 2021; 81: 3866-3876.e2.
[15]. Nacarelli T, Lau L, Fukumoto T, et al. NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol 2019; 21: 397–407.
[16]. Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Molecular Cell 2016; 61: 654–666.
[17]. Von Zglinicki T. Oxidative stress shortens telomeres. Trends in Biochemical Sciences 2002; 27: 339–344.
[18]. Sakamuri SSVP, Sure VN, Kolli L, et al. Glycolytic and Oxidative Phosphorylation Defects Precede the Development of Senescence in Primary Human Brain Microvascular Endothelial Cells. GeroScience 2022; 44: 1975–1994.
[19]. Miwa S, Jow H, Baty K, et al. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat Commun 2014; 5: 3837.
[20]. Ramírez-Camacho I, Flores-Herrera O, Zazueta C. The relevance of the supramolecular arrangements of the respiratory chain complexes in human diseases and aging. Mitochondrion 2019; 47: 266–272.
[21]. Chen H, Chan DC. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Human Molecular Genetics 2009; 18: R169–R176.
[22]. Mitochondria form contact sites with the nucleus to couple prosurvival retrograde response. SCIENCE ADVANCES 2020; 16.
[23]. Wilson EL, Metzakopian E. ER-mitochondria contact sites in neurodegeneration: genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ 2021; 28: 1804–1821.
[24]. Simon DK, Lin MT, Zheng L, et al. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson’s disease. Neurobiology of Aging 2004; 25: 71–81.
[25]. Trifunovic A, Hansson A, Wredenberg A, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA 2005; 102: 17993–17998.
[26]. Pinto M, Moraes CT. Mechanisms linking mtDNA damage and aging. Free Radical Biology and Medicine 2015; 85: 250–258.
[27]. Correia‐Melo C, Marques FD, Anderson R, et al. Mitochondria are required for pro‐ageing features of the senescent phenotype. The EMBO Journal 2016; 35: 724–742.
[28]. Rizza S, Cardaci S, Montagna C, et al. S -nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc Natl Acad Sci USA; 115. Epub ahead of print 10 April 2018. DOI: 10.1073/pnas.1722452115.
[29]. Johnson SC. Nutrient Sensing, Signaling and Ageing: The Role of IGF-1 and mTOR in Ageing and Age-Related Disease. In: Harris JR, Korolchuk VI (eds) Biochemistry and Cell Biology of Ageing: Part I Biomedical Science. Singapore: Springer Singapore, pp. 49–97.
[30]. Yang W, Nagasawa K, Münch C, et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 2016; 167: 985-1000.e21.
[31]. Camacho-Pereira J, Tarragó MG, Chini CCS, et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metabolism 2016; 23: 1127–1139.
[32]. Chini CCS, Peclat TR, Warner GM, et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nat Metab 2020; 2: 1284–1304.
[33]. Covarrubias AJ, Kale A, Perrone R, et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat Metab 2020; 2: 1265–1283.
[34]. Xu H, Guan N, Ren Y-L, et al. IP3R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol 2018; 19: 140.
[35]. Bravo‐Sagua R, Parra V, López‐Crisosto C, et al. Calcium Transport and Signaling in Mitochondria. In: Terjung R (ed) Comprehensive Physiology. Wiley, pp. 623–634.
[36]. Brookes PS, Yoon Y, Robotham JL, et al. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American Journal of Physiology-Cell Physiology 2004; 287: C817–C833.
[37]. Müller M, Ahumada-Castro U, Sanhueza M, et al. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated With Aging. Front Neurosci 2018; 12: 470.
[38]. Wiel C, Lallet-Daher H, Gitenay D, et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun 2014; 5: 3792.
[39]. McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. Journal of Cell Biology 2018; 217: 65–77.
[40]. Chapman J, Fielder E, Passos JF. Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Letters 2019; 593: 1566–1579.
[41]. Davalli P, Mitic T, Caporali A, et al. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxidative Medicine and Cellular Longevity 2016; 2016: 1–18.
[42]. Korolchuk VI, Miwa S, Carroll B, et al. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017; 21: 7–13.
[43]. Miquel J, Economos AC, Fleming J, et al. Mitochondrial role in cell aging. Experimental Gerontology 1980; 15: 575–591.
[44]. Rowland AA, Voeltz GK. Endoplasmic reticulum–mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 2012; 13: 607–615.
[45]. Poston CN, Krishnan SC, Bazemore-Walker CR. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). Journal of Proteomics 2013; 79: 219–230.
[46]. Ziegler DV, Martin N, Bernard D. Cellular senescence links mitochondria-ER contacts and aging. Commun Biol 2021; 4: 1323.
[47]. Cieri D, Vicario M, Giacomello M, et al. SPLICS: a split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ 2018; 25: 1131–1145.
[48]. Kalia R, Wang RY-R, Yusuf A, et al. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018; 558: 401–405.
[49]. Joseph A-M, Adhihetty PJ, Wawrzyniak NR, et al. Dysregulation of Mitochondrial Quality Control Processes Contribute to Sarcopenia in a Mouse Model of Premature Aging. PLoS ONE 2013; 8: e69327.
[50]. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and Aging. Cell 2011; 146: 682–695.
[51]. Rassow J, Dekker PJT, Van Wilpe S, et al. The preprotein translocase of the mitochondrial inner membrane: function and evolution. Journal of Molecular Biology 1999; 286: 105–120.
[52]. Poyton RO, McEwen JE. CROSSTALK BETWEEN NUCLEAR AND MITOCHONDRIAL GENOMES. Annu Rev Biochem 1996; 65: 563–607.
[53]. Eisenberg-Bord M, Zung N, Collado J, et al. Cnm1 mediates nucleus–mitochondria contact site formation in response to phospholipid levels. Journal of Cell Biology 2021; 220: e202104100.
[54]. Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. The Journal of Physiology 2014; 592: 829–839.
[55]. Liu T, Yang N, Sidor A, et al. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca 2+ Leak. Circ Res 2021; 128: 1191–1204.
[56]. Giacomello M, Pellegrini L. The coming of age of the mitochondria–ER contact: a matter of thickness. Cell Death Differ 2016; 23: 1417–1427.
[57]. Wiel C, Lallet-Daher H, Gitenay D, et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun 2014; 5: 3792.
[58]. Madreiter-Sokolowski CT, Thomas C, Ristow M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox Biology 2020; 36: 101678.
[59]. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and Aging. Cell 2011; 146: 682–695.
[60]. Park JT, Lee Y-S, Cho KA, et al. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Research Reviews 2018; 47: 176–182.
[61]. Simon DK, Lin MT, Zheng L, et al. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson’s disease. Neurobiology of Aging 2004; 25: 71–81.
[62]. Saki M, Prakash A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radical Biology and Medicine 2017; 107: 216–227.
[63]. Tranah GJ. Mitochondrial–nuclear epistasis: Implications for human aging and longevity. Ageing Research Reviews 2011; 10: 238–252.
[64]. McCay CM, Crowell MF, Maynard LA. The Effect of Retarded Growth Upon the Length of Life Span and Upon the Ultimate Body Size. The Journal of Nutrition 1935; 10: 63–79.
[65]. Merry BJ. Oxidative stress and mitochondrial function with aging - the effects of calorie restriction. Aging Cell 2004; 3: 7–12.
[66]. Kim Y, Triolo M, Hood DA. Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle. Oxidative Medicine and Cellular Longevity 2017; 2017: 1–16.
[67]. Hidalgo-Gutiérrez A, González-García P, Díaz-Casado ME, et al. Metabolic Targets of Coenzyme Q10 in Mitochondria. Antioxidants 2021; 10: 520.
[68]. KC S, Càrcamo JM, Golde DW. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Gluti) and confers mitochondrial protection against oxidative injury. FASEB j 2005; 19: 1657–1667.
[69]. Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. The American Journal of Clinical Nutrition 2011; 93: 884S-890S.