







1. Introduction
If the cause of death were to be categorized by disease, stroke would rank as leading cause of death and disability in China [1], and the third leading cause of death in the Western world [2]. Stroke refers to the occlusion or hemorrhage of blood vessels in the brain, leading to a compromise in cerebral blood flow, ultimately resulting in brain cell dysfunction or death [2]. Specifically, a blood clot or plaque can block a blood vessel, causing a rapid decline in blood flow and ATP levels in the ischemic region. This is followed by ionic imbalances and metabolic failure, potentially leading to cell death within minutes. Stroke entails immediate treatment to salvage the tissue damage.
The term stroke holds personal trauma for me, as I witnessed my grandmother suffer stroke two times, the second of which ultimately claimed her life. Thus, when I had the opportunity to engage in scientific research, my immediate focus was on investigating the genetic causes, pathogenic mechanisms, and potential treatments for stroke.
The major risk factors for stroke include hypertension, atherosclerosis, and genetic predisposition. Studies have shown that managing hypertension to achieve moderate blood pressure reductions can significantly reduce both the frequency and fatality of strokes [3]. Additionally, clinical trials have demonstrated that lipid-lowering drugs and treatments aimed at improving vascular function can significantly reduce coronary death rates and show promising effects in stroke prevention [4]. However, unlike other risk factors, the genetic basis of stroke does not follow Mendelian inheritance patterns, suggesting that the genetic factors contributing to stroke are complex [2].
Among various genetic risks, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), a monogenic disorder, markedly increases the risk of stroke. In addition to stroke, typical CADASIL symptoms include headaches, cognitive disabilities, dementia, and death [3]. Research has shown that CADASIL is caused by mutations in the Notch3 protein, and individuals carrying Notch3 variants have double the risk of stroke compared to non-carriers [4].
Most CADASIL-associated mutations involve the loss or addition of cysteine residues in exons 3 and 4, resulting in an abnormal number of cysteines and disrupting the formation of disulfide bonds. The free cysteines may interfere with the Notch3 signaling pathway, leading to the accumulation of the extracellular domain of Notch3 in small arteries [4], thereby triggering CADASIL. In vivo studies on mice have demonstrated that the accumulation of Notch3 is responsible for the loss of vascular smooth muscle cells (VSMCs) and irreversible vascular damage [5].
This study focuses on a prototypical CADASIL-associated mutation, R90C in the EGFR2 domain, which is one of the most prevalent mutations among CADASIL patients [6]. Currently, no effective therapies exist for CADASIL, and only empirical treatments are available to alleviate symptoms. Thus, the mutated Notch3 protein may serve as a potential therapeutic target for CADASIL [7].
This research employs computational approaches from cheminformatics and bioinformatics to identify and design small molecules that exhibit high binding affinity to the mutated Notch3 structure (R90C); these molecules could potentially serve as drug candidates. The workflow is as follows: First, drugs from the DrugBank database [8] are clustered to identify those with high binding affinity to the mutated Notch3 (R90C) protein, with binding affinity predicted using Autodock Vina (via the Swiss-Dock web server) [9]. Next, for the drug classes with high binding affinity, a similarity search is performed in the ChemBL database [10] to find additional small molecules with similar structures and high binding affinity. Finally, structural alignment is conducted on these high-affinity molecules to identify common substructures, and the chemical properties of these substructures are analyzed.
2. Materials and Methods
Considering the significance of the Notch3 R90C mutation in stroke mechanism, the primary target protein in this study is the mutated form of Notch3, specifically the R90C variant, where the 90th amino acid is mutated from arginine (R) to cysteine (C). However, since neither the native protein nor its mutant form has an experimentally resolved structure, we predicted the structure of the R90C mutant using AlphaFold [11, 12], the most advanced and accurate protein structure prediction tool available.
Given that the ultimate goal of this study is to identify potential drug candidates, we started with the DrugBank database. For approximately 2,000 validated small-molecule drugs in DrugBank, we first performed hierarchical clustering of all the small molecules and selected a ‘representative’ molecule from each cluster.
Next, we predicted the binding affinity of each representative molecule using Autodock Vina (via the SwissDock web server), and the molecule with the highest binding affinity was identified as the first-order target molecule. Based on this target molecule, iterative similarity searches were conducted in the ChemBL database. Binding affinity predictions were again performed using Autodock Vina, and the molecule with the highest affinity was identified as the nth-order target molecule, depending on the number of iterations of the similarity search. Ultimately, the molecule with the highest binding affinity will be identified as a potential drug candidate (Fig. 1).
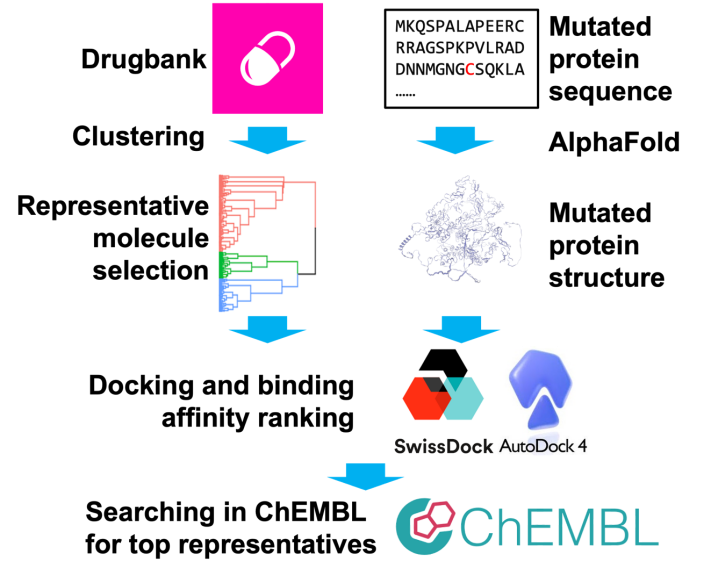
Figure 1. The pipeline for identifying potential drugs targeting Notch3 (R90C)
2.1. Obtaining the structure of the mutant
R90C [6], one of the most prevalent CADASIL-inducing mutations, is selected as the focus of this study. The sequence of the wild-type Notch3 protein is downloaded from the Uniprot database [13], and the sequence of the R90C mutant is obtained by manually modifying the wild-type sequence. The structure of the R90C mutant is then predicted using AlphaFold [11, 12] (Fig. 2).
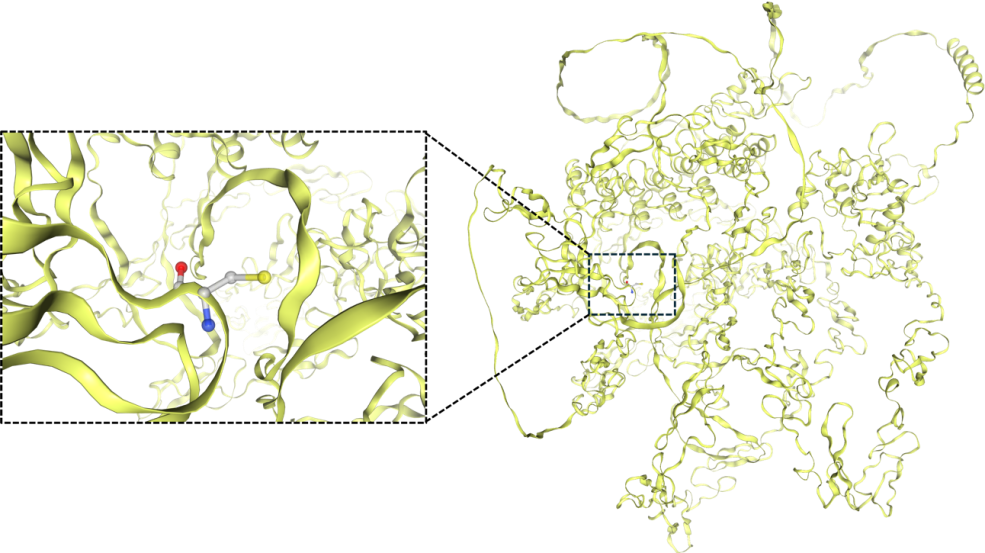
Figure 2. Predicted structure of the Notch3 mutant (R90C), with a zoom-in view of the R90C mutation site
2.2. Checking the conservation of the mutation site
To examine and detect the conservation at position 90 in the Notch3 protein, we used BlastP [14] to search for similar sequences in the Swiss-Prot database [15]. A multiple sequence alignment (MSA) was then performed on these sequences to ensure that the sites are aligned at the same position. Based on this alignment, we generated a WebLogo [16] plot, which is a graphical representation of sequence alignments that displays the conservation of amino acids at each position. The height of each letter in the plot reflects the frequency or conservation level of that residue. To highlight the conservation at the mutation site, we displayed only the first 140 positions of the WebLogo plot.
2.3. Identifying the binding pocket of the protein
Due to the requirement of selecting a binding search box (or binding site) in AutoDock Vina and the size restrictions of the seaching space of the Swissdock web server, binding could only be limited to a specific region of the Notch3 protein mutant (R90C), rather than the entire protein.
Therefore, two central regions were chosen for this purpose. One region is centered around the mutation site, as the mutation site is relatively enclosed; the other region is centered around a binding cavity identified using the CB-dock2 [17], which is a tool used to predict protein binding sites by analyzing the spatial features of binding cavities and the structural information of ligands. This binding cavity is located close to the mutation site in space (within 10 Å), and it may be a major binding cavity of the mutant (Fig. 3).
2.4. Impact of the mutation on protein function
To assess whether the R90C mutation in the Notch3 protein is deleterious or beneficial, we employ the DDmut method [18], a computational tool designed to predict the effects of single amino acid substitutions on protein stability and function. DDmut evaluates how specific mutations influence the protein's overall stability, binding affinity, and structural integrity.
The method begins by analyzing the wild-type Notch3 protein structure and comparing it to the structure of the R90C mutant. DDmut calculates the impact of the R90C mutation on the protein's stability by assessing changes in free energy associated with the mutation. It also considers potential disruptions to protein-protein interactions and the overall functional impact of the mutation.
By quantifying these effects, DDmut provides insights into whether the R90C mutation contributes to protein destabilization or functional impairment, which is crucial for understanding its role in diseases such as CADASIL. This analysis helps determine whether the mutation is likely to be harmful, leading to protein dysfunction and disease, or if it might have a neutral or even beneficial effect on the protein's function.
2.5. Clustering of small molecules in DrugBank database
To cluster the DrugBank dataset, small molecules in the Simplified Molecular Input Line Entry System (SMILES) format must first be converted into a more mathematically processable format—Morgan fingerprints [19]. Morgan fingerprints are bit vectors that capture the presence or absence of specific functional groups in substructures around atoms, and they are used to calculate molecular similarities. Using the Python toolkit RDKit [20], SMILES strings are converted into Morgan fingerprints, with each small molecule represented as a 2048-bit vector using RDKit's default parameters.
Agglomerative hierarchical clustering [21] is then applied to the resulting fingerprints based on Tanimoto similarity, which measures the structural similarity between two molecules by comparing the intersection and union of their features. In the context of Morgan fingerprints, Tanimoto similarity [22] \( T(A,B) \) , between fingerprints A and B is calculated as:
\( T(A,B)=\frac{|A∩B|}{|A|+|B|-|A∩B|} \) (1)
where \( |A| \) is the length of fingerprint \( A \) , \( |B| \) is the length of fingerprint \( B \) , and \( |A∩B| \) is the number of bits \( A \) and \( B \) share in common. Tanimoto index ranges from 0 to 1, where 0 indicates no overlap, and 1 indicates complete overlap between the two input molecules.
Agglomerative hierarchical clustering performs iteratively by merging the two nearest clusters of molecules into a larger cluster. The distance between two clusters is defined as one minus the average Tanimoto similarity between every pair of molecules across the two clusters. At the beginning, each molecule is considered an individual cluster. The algorithm continues until only one cluster remains, resulting in a dendrogram. Distinct clusters of molecules are identified by cutting the dendrogram at an empirically determined threshold, which stops the merging of clusters that exceed a certain distance. This thresholding ensures that the resulting clusters are distinct.
By classifying all small molecules from DrugBank into separate categories, the centroids of each cluster are identified as representatives of each drug molecule category.
2.6. Protein and small molecule binding
The SMILES structures of the drug representatives obtained through clustering are recorded. Binding affinities between these representatives and the target protein are then predicted using AutoDock Vina [23] via webserver SwissDock [9] web server with default parameters.
AutoDock Vina is a widely used molecular docking software that predicts the optimal binding modes of small molecules to proteins by evaluating various orientations and conformations based on scoring functions. The process involves preparing the protein and ligand structures, defining a grid box around the binding site, and docking the ligand into this space to assess binding affinity. By focusing on a well-defined search area, AutoDock Vina enhances the accuracy of these predictions. The results help identify which drug representatives might exhibit the highest affinity for the target protein, guiding further investigation and potential drug development.
2.7. Iteratively identify potential drugs
The representative with the highest predicted binding affinity is selected, and a similarity search is then conducted in the ChemBL [10] small molecule dataset using the ChemMine Tools web server [24]. Molecules with a similarity score greater than a predefined threshold (> 0.7) are selected for docking studies by Autodock Vina. Each selected molecule is docked into the target protein, and the one with the highest predicted affinity is chosen for additional docking iterations.
This iterative similarity search process aims to identify potential drug molecules by leveraging known high-affinity representatives to find similar compounds with potentially higher binding affinities. By performing multiple rounds of similarity searches, a sufficient number of high-affinity molecules are identified, which are considered as potential drug candidates. This approach enhances the likelihood of discovering effective new drugs, even if the exact nature of the optimal drug molecule is initially unknown.
2.8. Identify the maximum common Structure
In the identification of the Maximum Common Substructure (MCS) among a set of molecules, we employed the RDKit [20] cheminformatics software, which utilizes graph-based algorithms to detect the largest contiguous subgraph present across all molecules in the study. This is achieved through the rdFMCS.FindMCS() function, which compares graph representations of molecules where atoms are nodes and bonds are edges. The MCS search can be tailored by specifying match criteria for atoms and bonds, including atom elements and bond types. Additionally, options are provided to handle special cases such as ring matching. The outcome of the MCS search is provided as a SMILes string, representing the chemical structure common to all input molecules. This method is particularly useful in identifying a shared pharmacophore or scaffold, aiding in the optimization of drug-like properties.
3. Results
3.1. Notch3 protein and R90C variant
The Notch3 protein, specifically the R90C variant, is a key focus in this study. The original sequence of the Notch3 protein is sourced from the UniProt [13] database, under the entry Q61982. This sequence is characterized by its role in the regulation of vascular smooth muscle cells, and the R90C mutation is particularly notable due to its association with CADASIL, a hereditary stroke disorder. The mutation involves the substitution of arginine (R) at position 90 with cysteine (C), potentially altering the protein's function and stability.
To further investigate the structural implications of this mutation, the protein structure of the R90C variant is predicted using AlphaFold [11, 12], an advanced AI-based tool for protein structure prediction. AlphaFold generates high-accuracy models of the protein, allowing for detailed visualization and analysis of how the mutation affects the overall structure and potential binding sites. This structural information is crucial for understanding the functional consequences of the R90C mutation and its role in disease mechanisms.
To analyze the conservation of the Notch3 protein sequence, I used BlastP to search for similar protein sequences across various species. This allowed me to evaluate the conservation of specific amino acids, particularly around the R90C mutation. After retrieving homologous sequences, a web logo (Fig. 3) was generated to visualize sequence conservation, focusing on the first 140 amino acids of the protein.
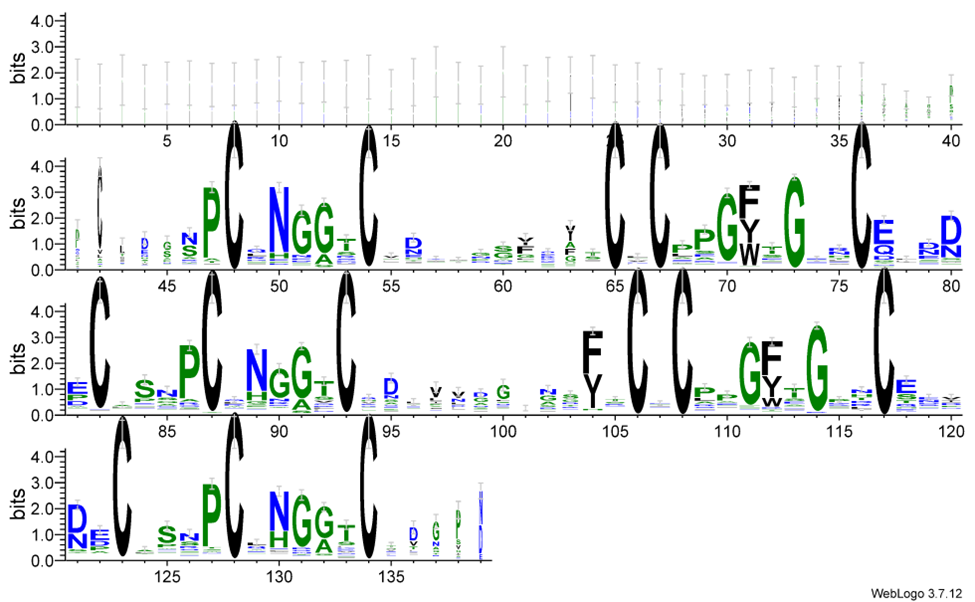
Figure 3. The WebLogo plot shows that R90C occurs in a non-conserved region.
The resulting WebLogo revealed that cysteines (shown as C in the WebLogo plot) are among the most highly conserved residues in this region. Cysteines play a crucial role in protein stability by forming disulfide bonds, which are essential for maintaining the protein's three-dimensional structure and facilitating proper folding. Given that the R90C mutation substitutes an arginine with a cysteine, this change is likely to disrupt disulfide bond formation. This disruption could lead to alterations in the protein's structure, affecting its function and potentially contributing to disease mechanisms such as those seen in CADASIL.
However, in the AlphaFold prediction of the R90C mutant, the cysteine at position 90 resulting from the mutation does not form new disulfide bonds with other cysteines, nor does it disrupt the existing disulfide bonds. This could be due to the existing disulfide bonds may exhibit significant thermodynamic stability, preventing disruption by the introduction of the new cysteine at position 90. Misfolding of the protein is therefore unlikely to occur under normal conditions, unless triggered by external factors such as changes in environmental conditions or specific molecular interactions that could destabilize the native disulfide architecture.
Given that the newly predicted Notch 3 protein mutant (R90C) does not disrupt existing disulfide bonds or form new ones, we utilize DDmut [18] to evaluate the impact of this mutation on the overall protein structure. DDmut is a computational tool designed to assess the effects of amino acid substitutions on protein stability and function by predicting changes in free energy. It calculates how specific mutations influence the protein's stability, binding affinity, and overall structural integrity. By applying DDmut, we can determine whether the R90C mutation affects the stability of the protein locally and how these changes might propagate throughout the protein structure, potentially altering its function and contributing to disease mechanisms.
From the results of DDmut (Fig. 4), the R90C mutation introduces a substitution at position 90, where the side chain of the newly added cysteine forms a new polar interaction (shown in orange in Fig. 5). Specifically, the nitrogen atom of the cysteine side chain at position 90 interacts with the oxygen atom at position 88. This new interaction results in a decrease in the overall free energy of the protein structure by 0.06 kcal/mol. This reduction in free energy contributes to a slight increase in structural stability, suggesting that the R90C mutation may enhance the stability of the protein by introducing favorable electrostatic interactions.
To prepare for the subsequent protein docking, where AutoDock Vina requires predefined binding centers and corresponding binding boxes, we need to identify these binding centers in advance. We employed two methods for this purpose: one approach uses the spatial position of the R90C mutation as the binding center, while the other utilizes the binding cavity predicted by the CB-dock2 [17] method. In fact, the binding centers identified by these two methods are not far apart, with a spatial distance of less than 10 Å (Fig. 5). Both binding sites will be used in the forthcoming docking studies along with appropriate binding boxes.
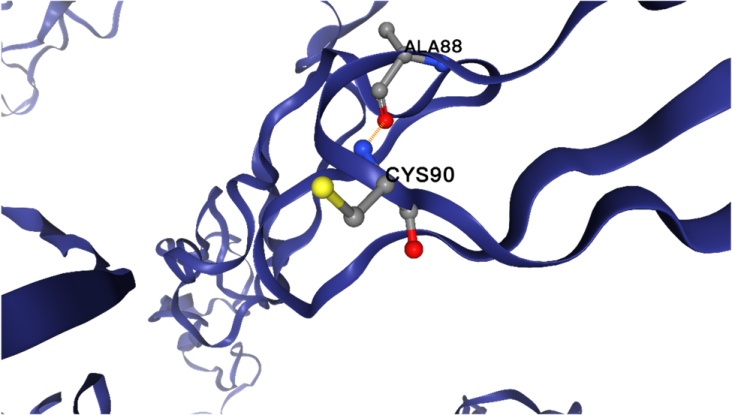
Figure 4. The R90C mutation slightly stabilizes the protein structure by introducing a new polar interaction.
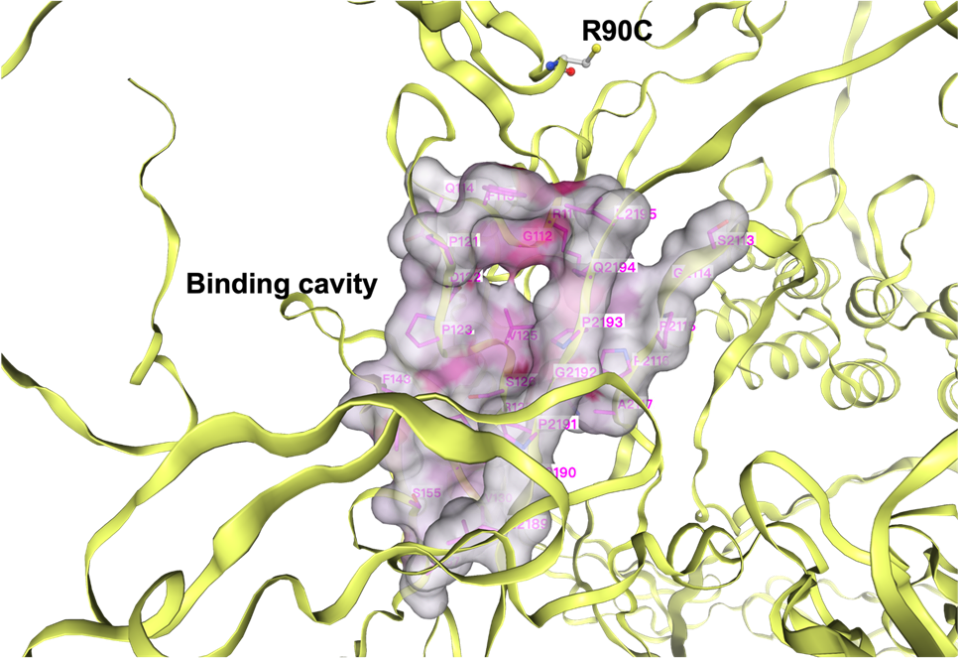
Figure 5. Two search centers: the binding cavity and the mutation site (R90C)
3.2. Screening and Classification of Small Molecule Drugs in DrugBank
We began our search for small molecule drugs using the DrugBank database, which provides comprehensive information about approved and experimental drugs. Among approximately 6,000 entries in DrugBank, we filtered out compounds that are not small molecules or do not have valid SMILES (Simplified Molecular Input Line Entry System) structures, which is a format used to represent chemical structures in a concise way. After this initial screening, we narrowed down the list to 2,681 small molecules.
Given the substantial number of candidates and our limited computational resources, we sought to reduce the computational load. We calculated the Tanimoto similarity between each pair of small molecules, which quantifies the similarity based on shared structural features. Using this similarity data as a distance metric, we applied hierarchical clustering to group the 2,681 small molecules into clusters. This method resulted in 18 distinct clusters (Fig. 6).
Among these 18 categories, we select the central small molecule structure from each classification as the "representative" of that category (Table 1). Many of these representatives include well-known drugs, such as Salicylic acid (C2), which is widely used as an anti-inflammatory and keratolytic agent, particularly in the treatment of skin conditions such as acne, psoriasis, and warts. It helps alleviate symptoms by reducing inflammation and excessive cell proliferation. Another example sulfamethoxazole (C12), which is an antimicrobial agent that, when combined with trimethoprim, forms a drug used to treat bacterial infections. It works by inhibiting bacterial folate synthesis, thereby preventing the growth and reproduction of bacteria.
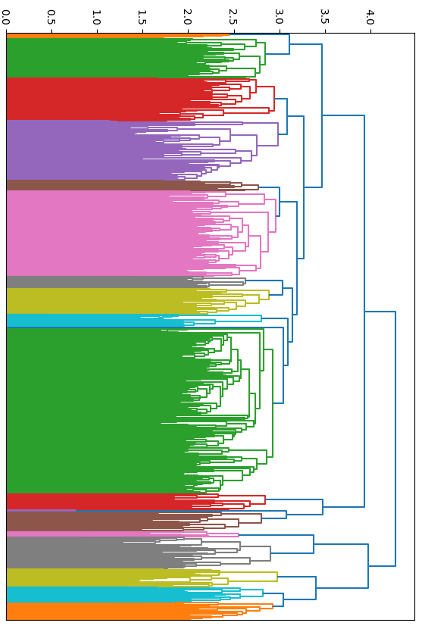
Figure 6. The 2,681 small molecule drugs from DrugBank were hierarchically clustered into 18 categories (from top to bottom: categories 1-18).
While we also considered using the K-means clustering algorithm, we chose hierarchical clustering because it does not require specifying the number of clusters in advance, thus avoiding the introduction of predefined parameters and providing a more flexible clustering approach.
Table 1. 18 representatives and their SMILES and common names
Clusters | Representative SMILES | Common name |
C1 | C1=CC=C(C=C1)CC(C(=O)O)N | N-Phenylglycine |
C2 | C1=CC=C(C=C1)C(=O)O | Salicylic Acid |
C3 | CCC(C)C(C(=O)NC(CC(C)C)C(=O)O)NC(=O)C(CC1=CC=C(C=C1)O)NC(=O)C2CCCN2C(=O)C(CCCCN)NC(=O)C(CCCCN)NC(=O)C(C(C)O)NC(=O)C(C)NC(=O)C(CC(=O)N)NC(=O)C(CO)NC(=O)CNC(=O)CNC(=O)C(CCC(=O)O)NC(=O)C(CCC(=O)O)NC(=O)C(CO)NC(=O)C3CCC(=O)N3 | Glycineamide Derivative |
C4 | CC12CCC(=O)C=C1CCC3C2C(CC4(C3CCC4(C(=O)CO)O)C)O | Bicyclic Lactone |
C5 | CC1=CC(=NC(=N1)NS(=O)(=O)C2=CC=C(C=C2)N)C | 4-Nitrophenylhydrazone |
C6 | CC(C)NCC(C1=CC(=C(C=C1)O)O)O | 3,4-Dihydroxyphenylacetic Acid |
C7 | CC(CC1=CC=CC=C1)N | N-Ethyl-2-pyrrolidinone |
C8 | CC(=O)OCC1=C(N2C(C(C2=O)NC(=O)C(C3=CC=CC=C3)N)SC1)C(=O)O | Glycolic Acid |
C9 | COC1=C(OC2CCCC2)C=C(C=C1)C1CNC(=O)C1 | Caffeine Derivative |
C10 | CCOC(=O)C1(CCN(CC1)C)C2=CC=CC=C2 | N-Benzylacetamide |
C11 | C(C1C(C(C(C(O1)OC2C(OC(C(C2O)O)O)CO)O)O)O)O | Sucralose |
C12 | CCC(=O)SCCNC(=O)CCNC(=O)C(C(C)(C)COP(=O)(O)OP(=O)(O)OCC1C(C(C(O1)N2C=NC3=C(N=CN=C32)N)O)OP(=O)(O)O)O | Sulfamethoxazole |
C13 | C1=NC2=C(N1C3C(C(C(O3)CO)O)O)N=C(NC2=O)N | Benzoyl peroxide |
C14 | CCCCCCCCCCCCC(=O)O | Stearic Acid |
C15 | C(CC(=O)O)C(C(=O)O)N | Lactic Acid |
C16 | C1C(C2C(O1)C(CO2)O[N+](=O)[O-])O | Phenylbutyrate |
C17 | CC(=O)C(C(COP(=O)(O)O)O)O | Glucose 6-Phosphate |
C18 | CCCCO | Ethylene Glycol |
3.3. The docking between drug small molecules and the protein.
After preparing the Notch3 R90C mutant protein and the corresponding small molecules, we next use AutoDock Vina [23] (via SwissDock) to assess the binding affinity between the 18 representatives of each small molecules clusters and the protein. Specifically, if a small molecule exhibits a lower binding affinity with the protein, it indicates a better interaction. This means that the small molecule has a higher likelihood of binding effectively and potentially protecting or affecting the cysteine residue introduced by the R90C mutation. Such small molecules, therefore, have the potential to be considered as promising drug candidates.
Table 2. AutoDock Vina binding affinities for the 18 representative small molecules and the two different binding sites on the protein.
Clusters | Binding affinity at R90C (kcal / mol) | Binding affinity at predicted cavity (kcal / mol) |
C1 | -4.289 | -4.937 |
C2 | -4.245 | -4.701 |
C3 | -5.348 | -5.402 |
C4 | -5.151 | -6.488 |
C5 | -4.360 | -4.701 |
C6 | -4.183 | -5.544 |
C7 | -5.849 | -4.932 |
C8 | -5.321 | -4.502 |
C9 | -4.721 | -6.440 |
C10 | -4.583 | -5.532 |
C11 | -4.657 | -4.739 |
C12 | -4.523 | -5.017 |
C13 | -5.031 | -5.428 |
C14 | -3.259 | -5.515 |
C15 | -3.516 | -3.928 |
C16 | -3.564 | -4.097 |
C17 | -3.799 | -3.976 |
C18 | -2.738 | -4.635 |
Table 2 illustrates the binding affinities of 18 small molecule representatives at two different binding sites on the Notch3 R90C mutant protein: the R90C mutation site and the predicted cavity. In the majority of cases, the predicted cavity shows stronger binding affinity (more negative values) compared to the R90C site. Specifically, 16 out of 18 clusters exhibit higher binding affinity at the cavity site. For instance, Cluster C4 shows a significant increase, with a binding affinity of -6.488 kcal/mol at the cavity, compared to -5.151 kcal/mol at the R90C site. Similarly, Cluster C9 has -6.440 kcal/mol at the cavity versus -4.721 kcal/mol at R90C.
On average, the predicted cavity consistently performs better as a binding site, reinforcing its potential as a more favorable target for drug interactions. However, in a few cases, the mutation site still shows comparable or even slightly better binding. For example, Cluster C7 has a higher affinity at the R90C site (-5.849 kcal/mol) compared to the cavity (-4.932 kcal/mol), indicating that while the predicted cavity is generally superior, the mutation site might still offer a viable binding option in select cases.
With the binding affinities of the representatives in hand, we can select the class with the optimal binding affinity and use ChemMine [24] to search for structurally similar small molecules within ChemBL [10]. For these small molecules, we then dock them with the R90C protein using AutoDock Vina to calculate their respective binding affinities. Specifically, at the mutation site (position 90), our chosen molecule is COC1=C(OC2CCCC2)C=C(C=C1)C1CNC(=O)C1 (commonly known as Caffeine Derivative, representing class C7). For the predicted cavity, the selected small molecule is CC12CCC(=O)C=C1CCC3C2C(CC4(C3CCC4(C(=O)CO)O)C)O, which has a binding affinity of -6.488 kcal/mol, representing class C4. This molecule shows the best binding affinity in both columns.
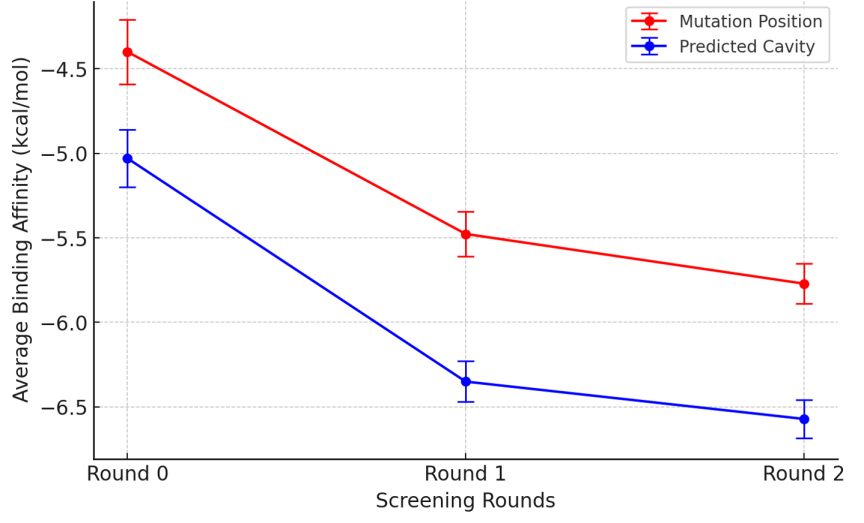
Figure 7. Comparsion of binding affinity across screening round.
Fig. 7 illustrates the binding affinities of selected small molecule candidates to both the mutation position and the predicted cavity across three screening rounds.
Round 0 represents the initial docking of the Notch3 protein with representatives from 18 different categories of small molecules. Round 1 involves selecting the category with the best binding affinity and identifying structurally similar small molecules from the ChemBL database. The average binding affinity and standard error are then calculated using these newly identified small molecules. Round 2 continues this process by selecting the small molecule with the highest binding affinity from the first round and conducting further searches. The top 10 small molecules identified in this search are used to compute the average binding affinity and standard error for this round. This sequential screening process progressively refines the selection of potential drug candidates by focusing on those with increasing binding affinities.
The significance of conducting multiple screening rounds is evident from the progression seen in the data. Each round refines the selection process, potentially yielding candidates with higher affinities and better therapeutic profiles. It also shows that the predicted cavity consistently outperforms the mutation position in terms of binding affinity across all rounds. This suggests that the predicted cavity offers a more favorable binding environment for these small molecules, indicating that focusing on this site may lead to the identification of more effective drug candidates.
Given these promising results, extending the screening to additional rounds could be beneficial. More rounds would allow for an even finer filtration of candidates, potentially leading to the discovery of small molecules with optimal binding characteristics. This iterative screening process, especially when paired with a focus on areas like the predicted cavity, represents a powerful strategy in the quest for novel therapeutic agents. This method not only enhances the efficiency of drug discovery but also improves the chances of finding highly specific and effective drugs.
3.4. The docking between drug small molecules and the protein.
After identifying a series of small molecules that effectively interact with the target protein, we further explored what factors enable these molecules to achieve good binding affinity with the Notch3 R90C mutant protein. Here we only used the round 2 screening as the example. We focused on identifying the largest common structural feature among these molecules, which was shown in Fig. 8. This structure, representing the maximum common substructure, was then exclusively used for docking with the target protein.
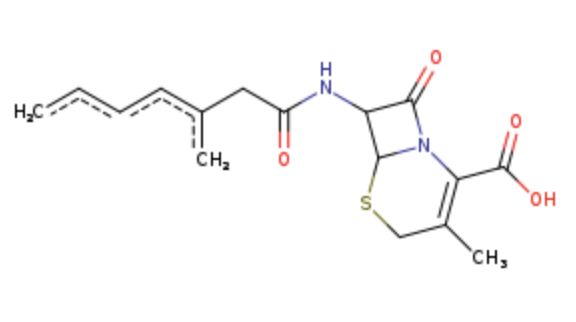
Figure 8. The largest common structure of round 2 screening on predicted cavity.
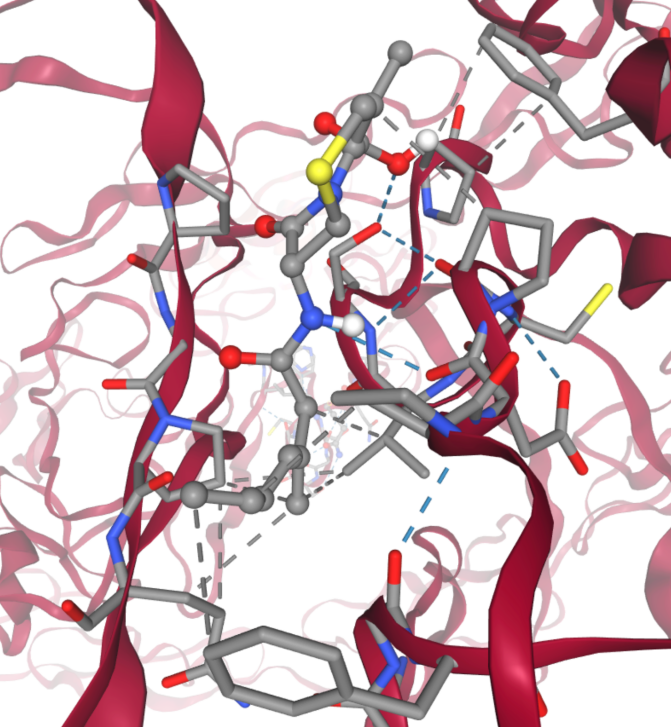
Figure 9. Hydrogen bond (blue) and hydrophobic contact (grey) enhance the binding between notch 3 mutant R90C and round 2 screened small molecules.
Through this targeted docking approach, we discovered that this common structural motif is capable of forming multiple hydrogen bonds with the protein's side chains (shown in blue in Fig. 9). Additionally, the structure establishes numerous hydrophobic contacts with the protein (shown in grey in Fig. 9), further enhancing the tightness of the binding. The presence of a cyclic component with both amide and thioether functionalities within the common structure suggests a unique combination of flexibility and rigidity, allowing optimal orientation and interaction within the protein’s binding site. This structural synergy likely contributes significantly to the observed high binding affinities, underscoring the potential of focusing on such common features in drug design efforts.
4. Discussion
The study presented utilized iterative screening to identify potential therapeutic agents for targeting the Notch3 R90C mutant, a protein variant linked to CADASIL. By applying a combination of molecular docking (AutoDock Vina) and hierarchical clustering within the DrugBank database, the research demonstrated the practical application of computational tools to enhance drug discovery processes. However, this approach, while innovative, also exposes several areas of potential improvement and challenge that warrant further exploration and discussion.
4.1. Limitations of DrugBank Screening
One notable limitation encountered in this study is the reliance on the DrugBank database, which, despite its extensive collection, provides a finite set of small molecules. The initial screening of these molecules, although systematic, may have inadvertently excluded potential candidates not listed in DrugBank or those not categorized as small molecules suitable for binding studies. Additionally, the classification of molecules in DrugBank, based largely on therapeutic categories rather than chemical properties or specific binding capabilities, may not have been sufficiently granular to capture the nuances required for targeting a specific protein mutation. This generalization potentially limits the discovery of novel or unexpected interactions that could be therapeutically relevant.
Another critical reflection on the utilization of the DrugBank database is that we simply applied hierarchical clustering to process the data from DrugBank, which does not fully leverage the advantages of the database.
4.2. Insufficiency of Iterative Screening Round.
The methodology employed only 2 rounds of screening. This limited number of rounds may not adequately represent the depth of screening necessary to conclusively identify the best candidates. Each round refines the pool based on the highest binding affinities, yet the variability and true potential of slightly lower-ranked molecules might not be fully explored. The cut-off for progression to subsequent rounds may thus exclude candidates with beneficial off-target effects or those that could exhibit improved efficacy upon slight molecular modification.
4.3. Reliance on Predictive Models
Another critical point is the exclusive use of AlphaFold-predicted structures for the docking studies. While AlphaFold represents a groundbreaking advancement in structural biology, providing highly accurate protein models, these are still predictions and not experimentally determined structures. The reliance on computational predictions carries inherent risks, such as inaccuracies in modeling dynamic protein conformations or interactions under physiological conditions. This reliance might skew the binding affinity data and interaction analyses, potentially leading to misleading conclusions about a molecule's therapeutic viability.
4.4. Simplification of Molecular Interactions
The study's approach to characterizing interactions primarily focused on binding affinities, which, while crucial, do not encompass the full spectrum of biochemical interactions relevant in a physiological context. Interactions such as allosteric effects, transient binding events, and the influence of cellular microenvironments were not accounted for. These factors can significantly affect a drug's performance and are often only observable in more complex biological assays or in vivo studies.
4.5. Potential for Overlooking Synergistic and Polypharmacological Effects
In focusing on single-target interactions, the study may overlook the potential synergistic effects that could arise from molecules affecting multiple pathways or targets. Polypharmacology, the design or use of drugs that affect multiple targets, is increasingly recognized as a valuable approach in treating complex diseases like CADASIL, which may involve multiple pathological pathways. The current methodology, by concentrating solely on the R90C mutation's direct binding partners, potentially misses opportunities to explore compounds that offer broader therapeutic effects.
4.6. Future Directions
To address these limitations, future research should consider expanding the chemical space explored through integration with additional databases and synthetic libraries to bypass the constraints posed by existing classifications. Increasing the number of screening rounds and incorporating feedback mechanisms to revisit excluded molecules could also enhance the comprehensiveness of the screening process. Moreover, integrating experimental validation stages early in the screening process would help confirm the computational predictions, thereby solidifying the basis for drug development decisions.
Additionally, adopting a systems biology approach to account for the complex network of interactions within cellular systems could provide a more holistic view of a drug candidate’s potential impact. Such an approach could also help identify multi-target drugs that may offer better therapeutic profiles for complex genetic disorders like CADASIL.
In conclusion, while the study sets a robust foundation for using computational methods in drug discovery, its insights must be further refined and expanded through integrated, multi-dimensional approaches to drug research and development. This will not only overcome the identified limitations but also pave the way for more effective and comprehensive therapeutic solutions.
5. Conclusion
The findings from this study underscore the potential of iterative screening methodologies to significantly enhance drug discovery efforts. By employing multiple screening rounds, we were able to progressively refine our pool of candidates, demonstrating that each subsequent round could indeed yield compounds with increasingly favorable binding characteristics. This process proved particularly effective in not only identifying high-affinity binders but also in understanding the structural basis of their interactions with the Notch3 R90C mutation. The use of such a targeted approach confirms the practicality of using advanced computational tools and clustering techniques to streamline the drug development pipeline, potentially leading to more effective treatments for diseases with complex genetic backgrounds like CADASIL.
References
[1]. Zhou, M., et al., Mortality, morbidity, and risk factors in China and its provinces, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. The Lancet, 2019. 394(10204): p. 1145-1158.
[2]. Lo, E.H., T. Dalkara, and M.A. Moskowitz, Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci, 2003. 4(5): p. 399-415.
[3]. Chabriat, H., et al., Cadasil. Lancet Neurol, 2009. 8(7): p. 643-53.
[4]. Wang, T., M. Baron, and D. Trump, An overview of Notch3 function in vascular smooth muscle cells. Prog Biophys Mol Biol, 2008. 96(1-3): p. 499-509.
[5]. Dupre, N., et al., Protein aggregates containing wild-type and mutant NOTCH3 are major drivers of arterial pathology in CADASIL. J Clin Invest, 2024. 134(8).
[6]. Lin, C., et al., Notch3 and its CADASIL mutants differentially regulate cellular phenotypes. Exp Ther Med, 2021. 21(2): p. 117.
[7]. Monet, M., et al., The archetypal R90C CADASIL-NOTCH3 mutation retains NOTCH3 function in vivo. Hum Mol Genet, 2007. 16(8): p. 982-92.
[8]. Knox, C., et al., DrugBank 6.0: the DrugBank Knowledgebase for 2024. Nucleic Acids Res, 2024. 52(D1): p. D1265-D1275.
[9]. Bugnon, M., et al., SwissDock 2024: major enhancements for small-molecule docking with Attracting Cavities and AutoDock Vina. Nucleic Acids Res, 2024. 52(W1): p. W324-W332.
[10]. Zdrazil, B., et al., The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res, 2024. 52(D1): p. D1180-D1192.
[11]. Tunyasuvunakool, K., et al., Highly accurate protein structure prediction for the human proteome. Nature, 2021. 596(7873): p. 590-596.
[12]. Jumper, J., et al., Highly accurate protein structure prediction with AlphaFold. Nature, 2021. 596(7873): p. 583-589.
[13]. The UniProt, C., UniProt: the universal protein knowledgebase. Nucleic Acids Res, 2017. 45(D1): p. D158-D169.
[14]. Jacob, A., et al., Mercury BLASTP: Accelerating Protein Sequence Alignment. ACM Trans Reconfigurable Technol Syst, 2008. 1(2): p. 9.
[15]. Boutet, E., et al., UniProtKB/Swiss-Prot. Methods Mol Biol, 2007. 406: p. 89-112.
[16]. Crooks, G.E., et al., WebLogo: a sequence logo generator. Genome Res, 2004. 14(6): p. 1188-90.
[17]. Liu, Y., et al., CB-Dock: a web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol Sin, 2020. 41(1): p. 138-144.
[18]. Zhou, Y., et al., DDMut: predicting effects of mutations on protein stability using deep learning. Nucleic Acids Res, 2023. 51(W1): p. W122-W128.
[19]. Ehiro, T., Descriptor generation from Morgan fingerprint using persistent homology. SAR QSAR Environ Res, 2024. 35(1): p. 31-51.
[20]. Lovric, M., J.M. Molero, and R. Kern, PySpark and RDKit: Moving towards Big Data in Cheminformatics. Mol Inform, 2019. 38(6): p. e1800082.
[21]. Han, W., et al., Clustering on hierarchical heterogeneous data with prior pairwise relationships. BMC Bioinformatics, 2024. 25(1): p. 40.
[22]. Chung, N.C., et al., Jaccard/Tanimoto similarity test and estimation methods for biological presence-absence data. BMC Bioinformatics, 2019. 20(Suppl 15): p. 644.
[23]. Eberhardt, J., et al., AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J Chem Inf Model, 2021. 61(8): p. 3891-3898.
[24]. Backman, T.W., Y. Cao, and T. Girke, ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res, 2011. 39(Web Server issue): p. W486-91.
Cite this article
Lu,L. (2024). Enhancing Drug Discovery through Iterative Screening and Advanced Computational Analysis of Notch3 R90C Mutations. Theoretical and Natural Science,64,114-128.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Zhou, M., et al., Mortality, morbidity, and risk factors in China and its provinces, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. The Lancet, 2019. 394(10204): p. 1145-1158.
[2]. Lo, E.H., T. Dalkara, and M.A. Moskowitz, Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci, 2003. 4(5): p. 399-415.
[3]. Chabriat, H., et al., Cadasil. Lancet Neurol, 2009. 8(7): p. 643-53.
[4]. Wang, T., M. Baron, and D. Trump, An overview of Notch3 function in vascular smooth muscle cells. Prog Biophys Mol Biol, 2008. 96(1-3): p. 499-509.
[5]. Dupre, N., et al., Protein aggregates containing wild-type and mutant NOTCH3 are major drivers of arterial pathology in CADASIL. J Clin Invest, 2024. 134(8).
[6]. Lin, C., et al., Notch3 and its CADASIL mutants differentially regulate cellular phenotypes. Exp Ther Med, 2021. 21(2): p. 117.
[7]. Monet, M., et al., The archetypal R90C CADASIL-NOTCH3 mutation retains NOTCH3 function in vivo. Hum Mol Genet, 2007. 16(8): p. 982-92.
[8]. Knox, C., et al., DrugBank 6.0: the DrugBank Knowledgebase for 2024. Nucleic Acids Res, 2024. 52(D1): p. D1265-D1275.
[9]. Bugnon, M., et al., SwissDock 2024: major enhancements for small-molecule docking with Attracting Cavities and AutoDock Vina. Nucleic Acids Res, 2024. 52(W1): p. W324-W332.
[10]. Zdrazil, B., et al., The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res, 2024. 52(D1): p. D1180-D1192.
[11]. Tunyasuvunakool, K., et al., Highly accurate protein structure prediction for the human proteome. Nature, 2021. 596(7873): p. 590-596.
[12]. Jumper, J., et al., Highly accurate protein structure prediction with AlphaFold. Nature, 2021. 596(7873): p. 583-589.
[13]. The UniProt, C., UniProt: the universal protein knowledgebase. Nucleic Acids Res, 2017. 45(D1): p. D158-D169.
[14]. Jacob, A., et al., Mercury BLASTP: Accelerating Protein Sequence Alignment. ACM Trans Reconfigurable Technol Syst, 2008. 1(2): p. 9.
[15]. Boutet, E., et al., UniProtKB/Swiss-Prot. Methods Mol Biol, 2007. 406: p. 89-112.
[16]. Crooks, G.E., et al., WebLogo: a sequence logo generator. Genome Res, 2004. 14(6): p. 1188-90.
[17]. Liu, Y., et al., CB-Dock: a web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol Sin, 2020. 41(1): p. 138-144.
[18]. Zhou, Y., et al., DDMut: predicting effects of mutations on protein stability using deep learning. Nucleic Acids Res, 2023. 51(W1): p. W122-W128.
[19]. Ehiro, T., Descriptor generation from Morgan fingerprint using persistent homology. SAR QSAR Environ Res, 2024. 35(1): p. 31-51.
[20]. Lovric, M., J.M. Molero, and R. Kern, PySpark and RDKit: Moving towards Big Data in Cheminformatics. Mol Inform, 2019. 38(6): p. e1800082.
[21]. Han, W., et al., Clustering on hierarchical heterogeneous data with prior pairwise relationships. BMC Bioinformatics, 2024. 25(1): p. 40.
[22]. Chung, N.C., et al., Jaccard/Tanimoto similarity test and estimation methods for biological presence-absence data. BMC Bioinformatics, 2019. 20(Suppl 15): p. 644.
[23]. Eberhardt, J., et al., AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J Chem Inf Model, 2021. 61(8): p. 3891-3898.
[24]. Backman, T.W., Y. Cao, and T. Girke, ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res, 2011. 39(Web Server issue): p. W486-91.