







1. Introduction
The evolution of computational biology has been significantly influenced by the rapid progression in machine learning technologies, particularly with the advent of sophisticated models designed to tackle the complex nature of protein research. ProteinBERT, a transformer-based deep learning algorithm, stands out among these innovations. It has been specifically developed to recognize intricate patterns within protein sequences that traditional computational methods might overlook. This capability positions ProteinBERT as a pivotal tool in addressing fundamental questions in protein science, especially given its versatility across various protein-related challenges.ProteinBERT's application spans several critical areas of protein research. Antimicrobial peptides (AMPs), which are key components of the immune system, offer a broad spectrum of actions against microbes, making their classification vital for therapeutic advancements. Similarly, intrinsically disordered proteins (IDPs) challenge conventional structure-based protein prediction models due to their lack of stable three-dimensional structures, necessitating more advanced approaches like those offered by ProteinBERT for accurate analysis. Moreover, protein toxicity prediction remains essential for ensuring the safety and efficacy of therapeutic proteins, where understanding adverse effects is crucial. ProteinBERT's ability to navigate these complex areas reflects its integral role in advancing protein science.
This paper aims to provide a thorough analysis of ProteinBERT’s application across three domains: antimicrobial peptide classification, prediction of intrinsically disordered proteins, and protein toxicity assessment. By integrating domain-specific datasets and utilizing refined evaluation metrics, the effectiveness of ProteinBERT in these areas is rigorously investigated. This study not only assesses the current capabilities of ProteinBERT in enhancing the accuracy of predictions and classifications in protein research but also discusses potential improvements that could further optimize its performance. Moreover, the paper explores the broader implications of ProteinBERT's technology in computational biology, offering insights into its benefits and limitations while proposing future directions for research to expand its application and enhance its utility in the field.
2. Theoretical Foundations
2.1. The base model
The ProteinBERT model is a deep learning framework that has been specifically designed for protein sequences. It builds on the BERT (Bidirectional Encoder Representations from Transformers) architecture, initially developed for natural language understanding. The BERT architecture utilizes bidirectional attention, enabling the model to capture intricate dependencies between tokens in a sequence by assigning different attention weights to each token [1]. In natural language processing (NLP), this allows for the modeling of complex semantic relationships within a sentence. Similarly, in the context of protein sequences, ProteinBERT treats amino acids as tokens, thereby enabling the model to learn the complex relationships between residues across the entire sequence.
ProteinBERT has been trained on an extensive corpus comprising approximately 106 million protein sequences from the UniProtKB/ UniRef90 dataset, which encompasses a comprehensive range of known protein sequences. The pretraining process employs a dual-task approach, comprising bidirectional masked language modelling and Gene Ontology (GO) annotation prediction. Random tokens within the sequence are masked in the masked language modeling tasks, and the model is trained to predict these masked tokens based on the surrounding context. The GO annotation prediction task entails the prediction of protein functions, the capturing of diverse biological processes and molecular functions [2]. The combined pretraining strategy enables ProteinBERT to develop a comprehensive understanding of both the sequence structure and function, significantly enhancing its efficacy for downstream tasks such as antimicrobial peptide classification, intrinsically disordered protein prediction, and protein toxicity prediction.
Furthermore, ProteinBERT's architectural design incorporates several innovative features that differentiate it from the original BERT model and other protein language models. Notably, ProteinBERT distinguishes between local (character-level) and global (whole-sequence-level) representations, enabling more efficient and effective processing of long sequences. The model's architectural design, which incorporates convolutional and global attention layers, ensures that it is capable of handling sequences of varying lengths without the quadratic memory and computation growth that is associated with traditional self-attention mechanisms. This flexibility enables ProteinBERT to generalise effectively across a range of sequence lengths, making it particularly well-suited to tasks involving extremely long protein sequences [3].
ProteinBERT offers an efficient and scalable framework for protein sequence analysis, delivering near top-tier performance on multiple benchmarks while being smaller and faster than many competing models. The model's capacity to rapidly adapt to a range of protein-related tasks with minimal labelled data makes it a valuable tool in the field of bioinformatics.
2.2. Transformer layers
Transformer layers are the core components of ProteinBERT, consisting of multi-head self-attention mechanisms and feedforward neural networks. The attention mechanism operates by calculating an attention score for each pair of tokens in the sequence. The score thus serves to determine the extent to which one token should be the focus of attention when processing the sequence. Following the embedding process, the three vectors Q, K and V are generated by multiplying the word vectors with the three matrices \( {W^{Q}} \) , \( {W^{K}} \) and \( {W^{V}} \) . The dot product of Q and K represents the attention score between the two tokens in question. This is determined by calculating the dot product between the query vector of one token and the key vector of another. This assesses the degree of alignment between the current token's query and the other token's key, thus determining the extent to which the two are aligned. Subsequently, the scaling process is as follows: The dot product is often scaled by dividing it by the square root of the dimensionality of the key vectors. This step serves to stabilize the gradients during the training process, particularly when working with high-dimensional vectors. Subsequently, the resulting scores are subjected to a softmax function, which converts them into a probability distribution. This step ensures that all attention scores for a given token are equal, thereby determining the extent to which each token should focus on the others.
\( Attention(Q,K,V)=softmax(\frac{Q{K^{T}}}{\sqrt[]{{d_{k}}}})V \) (1)
In the traditional Natural Language Processing (NLP) tasks, self-attention performs by contracting the dependency relationship of each word in the sentence and assigning different relationship weights to each word, the different words in the context can be linked together to form the overall semantics. While in the protein region, the self-attention mechanism, enhanced by GO annotation, allows the model to evaluate the importance of individual amino acids within the sequence when making predictions, effectively capturing both short-range and long-range dependencies. The processing of the input sequence is conducted in parallel by each transformer layer, thereby enabling efficient computation and scalability to long sequences.
In practice, transformers like ProteinBERT use multi-head attention, where multiple sets of \( {W^{Q}} \) , \( {W^{K}} \) , and \( {W^{V}} \) matrices are learned. Each "head" in multi-head attention computes attention using a different set of learned \( {W^{Q}} \) , \( {W^{K}} \) , and \( {W^{V}} \) matrices. The outputs from all attention heads are concatenated and then processed through a linear layer to produce the final output.
The multi-head aspect of the self-attention mechanism enables ProteinBERT to learn different types of relationships between residues in parallel. Each head focuses on a distinct aspect of the sequence, such as specific motifs or structural patterns, thereby facilitating a more nuanced comprehension of the sequence [4]. Subsequently, the output is conveyed through a feedforward neural network, followed by layer normalization and residual connections. This facilitates the stabilization of the training process and enhances the model's capacity for generalization.
2.3. Local and global representations
A principal characteristic of ProteinBERT is its capacity to learn both local and global representations of protein sequences. Two nearly parallel paths make up the model architecture: one is for local representations and the other is for global representations. The global representations are 2D tensors with a shape of \( B×L×{d_{local}} \) , while the local representations are 3D tensors with a shape of \( B×L×{d_{local}} \) , where \( L \) is the minibatch sequence length, \( B \) is the batch size, and \( {d_{local}} \) is the number of channels [5]. Local representations capture the immediate context around each residue, which is essential for understanding short-range interactions and motifs that are critical for the function of antimicrobial peptides and the structural disorder in proteins. Such representations are of particular significance in tasks where specific subsequences or motifs are responsible for determining the overall function or property of the protein.
Conversely, global representations consider the entire sequence, capturing long-range dependencies and overall sequence characteristics that may influence protein toxicity or overall structure. This comprehensive approach is vital for tasks where the interaction between distant residues or the overall sequence composition is crucial for accurate predictions.
The architecture of ProteinBERT is designed to achieve a balance between local and global perspectives, thereby providing a robust framework for analysing protein sequences across a range of tasks. By employing both types of representations, ProteinBERT can achieve a high level of accuracy in complex prediction tasks, thereby establishing itself as a versatile tool in the field of computational biology. As show in the figure 1.
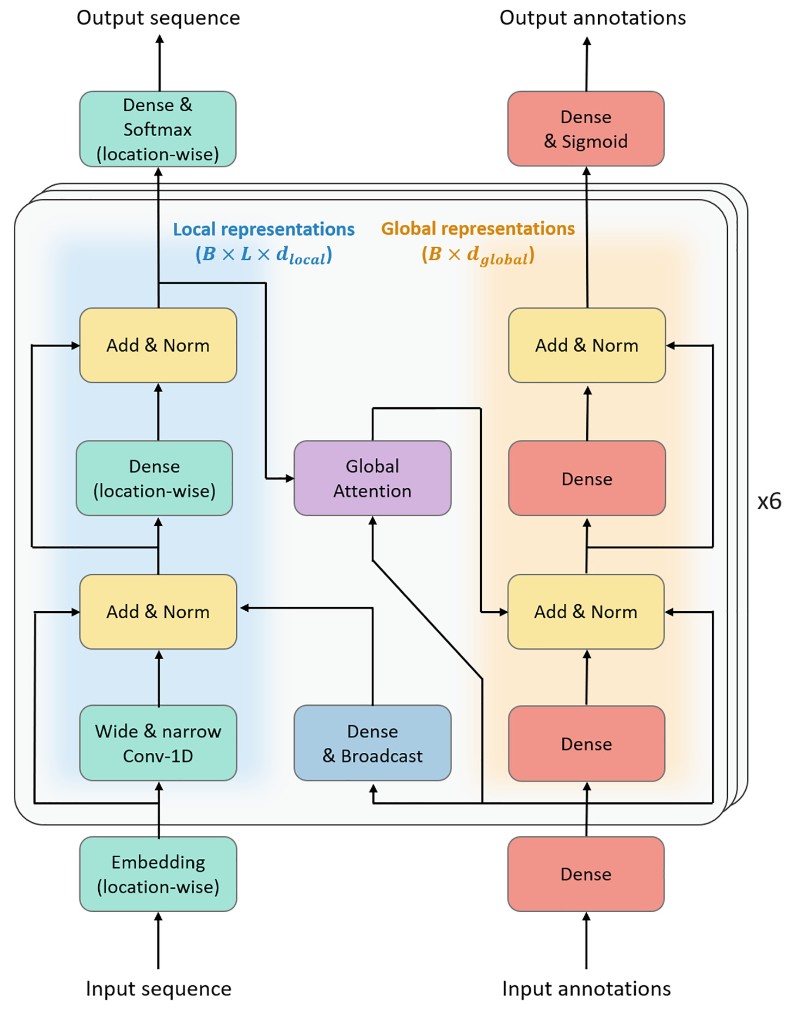
Figure 1. Neural network structure diagram [6].
3. Algorithm Analysis and Application Research
3.1. Classification of antimicrobial peptides
Antimicrobial peptides (AMPs) are small molecules, typically comprising 6 to 100 amino acids, which exhibit potent antibacterial properties. In contrast to conventional antibiotics, AMPs do not facilitate the development of bacterial resistance, thereby representing a promising avenue for the advancement of novel antibacterial therapeutics. The classification of AMPs is of great importance for the identification of potential therapeutic peptides [6]. To improve this task's accuracy and efficiency, a lot of research has been done recently on machine learning and deep learning techniques.
Conventional machine learning techniques, such as random forests and support vector machines, have been utilized for AMP classification. However, these approaches are subject to several limitations, including the necessity for extensive feature engineering, high computational costs and relatively low prediction accuracy. Feature engineering is a time-consuming process that requires significant domain expertise to transform raw data into informative features. The quality of the features that are engineered into these devices is a major factor in their effectiveness.
Recent studies have addressed similar tasks by using deep learning algorithms, which can automatically create representations of features from raw sequence data without requiring manual feature engineering. Among these, ProteinBERT model has demonstrated considerable potential in the classification of AMPs. ProteinBERT applies a pretraining-fine-tuning strategy, where the model is trained on a huge corpus of protein sequences to produce universal protein sequence representations, and then it is fine-tuned specific tasks like AMP classification.
The architecture of ProteinBERT for AMP classification comprises two parallel pathways: one for local sequence information and another for global sequence information. Both pathways are learned through self-supervised pretraining. The local pathway is responsible for capturing the detailed sequential information of amino acids, whereas the global pathway is tasked with capturing broader, contextual information across the entire protein sequence. During the process of fine-tuning, the model is optimised for the specific task of AMP classification, utilising the knowledge acquired during the pretraining phase to enhance the accuracy of the predictions [7].
ProteinBERT outperformed other deep learning models and conventional machine learning techniques in experimental tests. For example, in an evaluation of test sets, the ProteinBERT-based model achieved an accuracy of 92.35%, a sensitivity (SENS) of 92.70%, and a receiver operating characteristic (ROC) curve area of 97.3% [8]. These results not only exceed those of traditional methods but also surpass those of other state-of-the-art AMP classification models, including DNN and iAMPpred. The elevated accuracy and auROC value demonstrate that ProteinBERT is highly efficacious in differentiating between AMPs and non-AMPs, thereby establishing it as a reliable instrument for the identification of novel antimicrobial peptides.
Applying ProteinBERT to AMP classification marks a significant advancement in the field of bioinformatics. By leveraging deep transfer learning and attention mechanisms, ProteinBERT offers a more efficient and accurate approach to AMP classification, with the potential to accelerate the discovery of new antimicrobial agents.
3.2. Prediction of intrinsically disordered proteins
Intrinsically disordered proteins (IDPs) represent a distinctive and complex category within the field of proteomics, characterised by the absence of a fixed three-dimensional structure. This contrasts with the well-defined folds observed in globular proteins. The intrinsic disorder of IDPs allows them to adopt a multitude of conformations, which are essential for several biological processes, including cellular signalling, molecular transport and assembly. Because of these proteins' ability to dynamically interact with other cellular components, they have been connected to a number of neurodegenerative illnesses, such as Parkinson's and Alzheimer's [9].
The inherent flexibility and the limited availability of structured data have presented significant challenges for traditional machine learning and deep learning models in predicting the behaviour and structure of IDPs. However, ProteinBERT has been adapted to address this challenge. The architecture of ProteinBERT has been enhanced to improve its predictive capabilities for IDPs by incorporating a comprehensive dataset of IDP sequences. This enhancement enables ProteinBERT to predict a range of characteristics associated with IDP behaviour, including folding classes, secondary structures and remote homology.
The enhanced version of ProteinBERT was fine-tuned using a dataset comprising approximately 5,000 proteins, which were selected based on their intrinsic disorder characteristics. The dataset was meticulously cleaned and labelled for secondary structure, folding class, and remote homology, which are critical for understanding the diverse conformations that IDPs can assume [10]. Subsequently, the model was trained to predict these features across the IDP dataset, resulting in notable enhancements in accuracy and prediction capabilities.
In terms of performance, the fine-tuned ProteinBERT model demonstrated significant enhancements. Prior to the incorporation of data specific to IDPs, the model exhibited an accuracy of approximately 88% in folding class predictions and 74% in secondary structure predictions for standard proteins. However, the initial predictions for IDP data exhibited a notable decline in accuracy, particularly in the case of remote homology, where the accuracy rate approached zero. Following fine-tuning with the IDP dataset, the accuracy for folding class prediction among IDPs increased by 20%, reaching 93.97%. Similarly, an improvement of 6.768% was observed in the accuracy of secondary structure prediction, which demonstrates the enhanced capability of the model to handle the dynamic nature of IDPs [11].
Moreover, the predictions made by ProteinBERT for IDPs were analysed in terms of their distribution across different folding classes and secondary structures. The model predicted that a substantial proportion of IDPs (approximately 38.9%) belonged to the alpha and beta protein class, with an additional 26.99% belonging to the alpha protein class. This distribution is indicative of the model's capacity to discern and categorise the flexible structures of IDPs. Furthermore, the model accurately predicted amino acid sequences for approximately 98% of the IDP dataset, thereby further underscoring its robustness.
The incorporation of data specific to intrinsically disordered proteins into ProteinBERT signifies a substantial advancement in the prediction of such proteins. Researchers have improved the model's capacity to predict the intricate behaviors and structures of these proteins by fine-tuning it with an extensive dataset specifically designed for IDPs. This has made the model an invaluable tool for proteomics research. This development highlights the potential of machine learning in advancing our comprehension of IDPs, which have traditionally been challenging to study using conventional methodologies.
3.3. Prediction of protein toxicity
The prediction of protein toxicity represents a pivotal stage in the advancement of biologics, as it facilitates the identification of potentially deleterious proteins prior to their progression to clinical trials. While small molecules have established rules and predictive algorithms to assess toxicity, the prediction of toxicity in peptides and proteins has traditionally been less developed, resulting in higher failure rates during the later stages of drug development. A distinctive in silico protein toxicity classifier called CSM-Toxin was created to solve this problem. It predicts protein toxicity based exclusively on amino acid sequences by utilizing the ProteinBERT model.
Protein sequences are treated as sentences and amino acids like words in proteinBERT's BERT-based architecture, upon which CSM-Toxin is based. Since the model had been pre-trained using the Masked Language Model approach on more than 100,000 protein sequences from the database of UniProt, it was able to understand intricate connections between amino acids and their surrounding environment. Attribute to the pre-training, the model can make robust predictions even with minimal input features, relying entirely on the raw amino acid sequence to determine toxicity. As show in the figure 2.
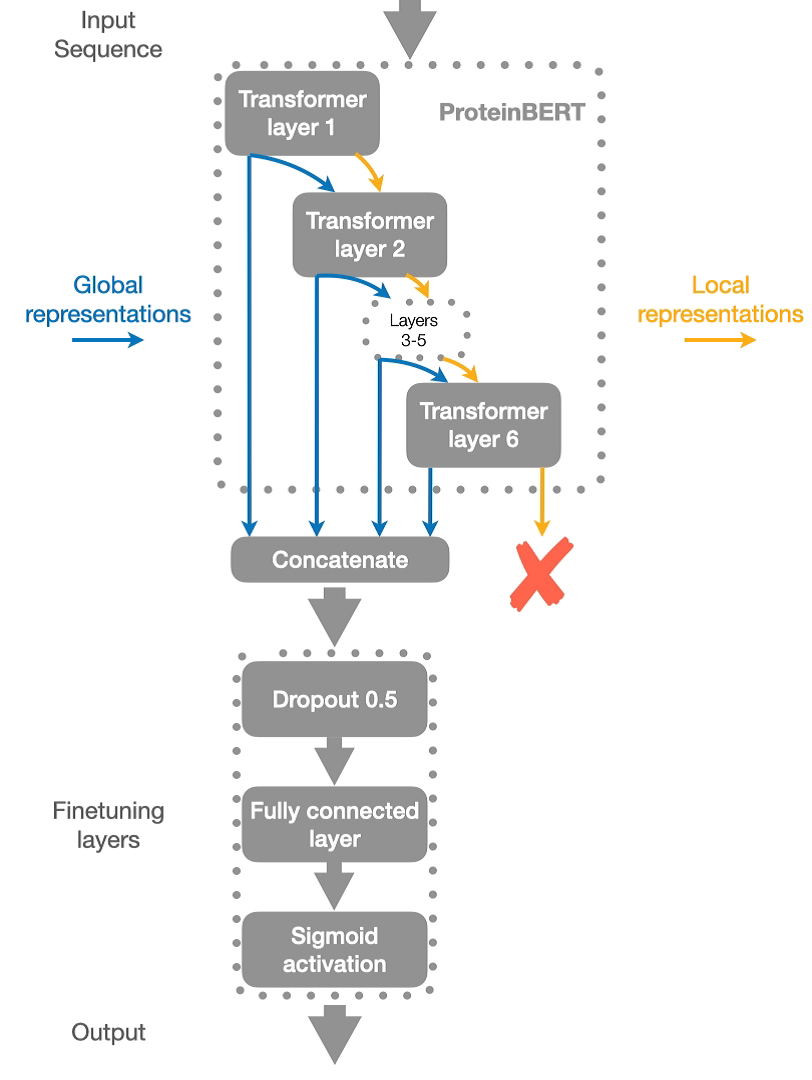
Figure 2. Structure diagram [12].
To develop CSM-Toxin, the researchers created the largest and most comprehensive dataset of experimentally measured toxic and non-toxic protein sequences. There were 214,740 non-toxic sequences and 2475 hazardous sequences in all. Carefully analyzing the dataset ensured that there were no duplicates and that the model could be used to a variety of protein types. Subsequently, the model was fine-tuned on this dataset, with a particular focus on optimising hyperparameters to enhance predictive performance. The application of cross-validation during the training phase revealed that CSM-Toxin attained 0.66 for Matthews Correlation Coefficient of and 0.86 for Area Under the Curve, thereby exhibiting superior performance in terms of both accuracy and robustness when compared to alternative methods.
CSM-Toxin consistently scored admirably in blind test assessments, obtaining an MCC of 0.64 and an AUC of 0.86 on a non-redundant test set. These results are noteworthy in that they indicate that the model is not only accurate but also consistent across different datasets, which is essential for practical applications in drug development. In comparison to alternative toxicity prediction models, such as ToxinPred2, CSM-Toxin exhibited enhanced precision, resulting in a reduction in the number of false positives while preserving a high recall level. In the case of toxicity prediction, where false positives and false negatives can both have serious consequences, striking an equilibrium between recall and precision is crucial.
The efficacy of ultilizing deep learning models like ProteinBERT for protein toxicity prediction is demonstrated by the accomplishments of CSM-Toxin. By focusing on the sequence alone, without the need for additional structural or evolutionary information, CSM-Toxin offers a streamlined and efficient approach to toxicity prediction. The model is accessible via a web server, thus enhancing its accessibility to researchers, who can rapidly assess the toxicity of protein sequences, thereby reducing the time and cost associated with biologic development.
4. Challenges and Limitations
Despite the significant progress achieved by ProteinBERT in various areas of protein research, several challenges and limitations remain that need to be addressed to further improve its applicability and accuracy.
One of the main challenges encountered in the application of ProteinBERT is the issue of data imbalance, particularly in tasks such as protein toxicity prediction. The curated datasets often contain a disproportionate number of non-toxic proteins compared to toxic proteins, which can lead to biased models that may underperform in identifying rare toxic proteins. In addition, the limited availability of experimentally validated datasets is a significant limitation. Prediction accuracy could be further improved with access to more comprehensive and balanced datasets covering a wider range of protein sequences and properties.
While ProteinBERT has shown strong performance on specific tasks after fine-tuning, its ability to generalise to entirely new protein sequences or unseen tasks remains a challenge. The model's performance, particularly in IDP and toxicity prediction, may deteriorate when applied to novel protein sequences that differ significantly from those in the training dataset. This limitation highlights the need for further improvements in model architecture and training strategies to improve ProteinBERT's ability to generalise to diverse and previously unseen protein sequences.
ProteinBERT requires significant computational resources to train and fine-tune, especially on large datasets. The model's complex architecture, which includes multiple layers of transformations and attention mechanisms, requires high memory and processing power, which may limit its accessibility to research groups with limited computational infrastructure. Although the model is efficient compared to other deep learning frameworks, the computational cost associated with training and deploying ProteinBERT at scale remains a notable limitation.
Another significant limitation is the interpretability of ProteinBERT's predictions. Although powerful, the model often operates as a 'black box', providing little insight into how specific predictions are made. This lack of interpretability can be particularly problematic in critical areas such as drug development and toxicity assessment, where understanding the rationale behind a prediction is critical. Improving the interpretability of ProteinBERT predictions through model explanatory techniques is an area that requires further exploration.
In addition, the quality of ProteinBERT’s pre-training on large protein sequence datasets has a significant impact on the performance in various tasks. Any biases or limitations in these pre-trained models may propagate into the fine-tuned models, potentially affecting their performance on specific tasks. This reliance on pre-trained models limits the flexibility to fully tailor the model architecture to specific tasks without risking losing the benefits gained from pre-training.
In summary, while ProteinBERT has made significant strides in advancing protein sequence analysis, addressing these challenges and limitations will be critical to its continued success and wider adoption in the field of bioinformatics. Future research should prioritize improving data availability and balance, improving generalization and interpretability of the model, and lowering the model's computational cost to fully realize its potential.
5. Conclusion
The implementation of ProteinBERT across various aspects of protein research marks a significant advancement in the field of bioinformatics. This study has validated the robustness and adaptability of ProteinBERT, showcasing its ability to accurately perform tasks such as the classification of antimicrobial peptides, prediction of intrinsically disordered proteins, and assessment of protein toxicity. Leveraging advanced mechanisms like deep transfer learning and attention-based models, ProteinBERT has outperformed traditional computational methods, offering enhanced accuracy and scalability. The fine-tuning of the model using domain-specific datasets has notably improved its efficacy in tackling complex predictions, underscoring its potential as a pivotal resource in proteomics and therapeutic development.
Further refinement of ProteinBERT is essential to fully harness its capabilities. Future research should aim to enhance the model's accuracy and efficiency, broadening its application scope to include a wider array of protein-related tasks. Additionally, there is a compelling opportunity to integrate ProteinBERT with other computational tools and platforms, which could amplify its utility across the broader spectrum of computational biology. Such integration could facilitate more comprehensive analyses and foster the development of innovative approaches in protein research, potentially accelerating discoveries in disease mechanisms and drug development.
References
[1]. Brandes, N., Ofer, D., Peleg, Y., Rappoport, N., & Linial, M. (2022). ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics, 38(8), 2102–2110.
[2]. Gene Ontology Consortium. (2004). The Gene Ontology (GO) database and informatics resource. Nucleic acids research, 32(suppl_1), D258-D261.
[3]. Oldfield, C. J., & Dunker, A. K. (2014). Intrinsically disordered proteins and intrinsically disordered protein regions. Annual review of biochemistry, 83(1), 553-584.
[4]. Jain, A., & Kihara, D. (2019). NNTox: Gene Ontology-Based Protein Toxicity Prediction Using Neural Network. Scientific Reports, 9(1), 17923.
[5]. Lu, J., Zhang, H., Jin, C., & Quan, X. (2023). A Novel Classification Method for Antimicrobial Peptides Based on ProteinBERT. 2023 42nd Chinese Control Conference (CCC), 8437–8442.
[6]. Brandes, N., Ofer, D., Peleg, Y., Rappoport, N., & Linial, M. (2022). ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics, 38(8), 2102-2110.
[7]. Zhu, X., Guo, C., Feng, H., Huang, Y., Feng, Y., Wang, X., & Wang, R. (2024). A Review of Key Technologies for Emotion Analysis Using Multimodal Information. Cognitive Computation, 1-27.
[8]. Wang R., Zhu J., Wang S., Wang T., Huang J., Zhu X. Multi-modal emotion recognition using tensor decomposition fusion and self-supervised multi-tasking. International Journal of Multimedia Information Retrieval, 2024, 13(4): 39.
[9]. Stone, J. D., Tabrizi, H. B., & Shamsuddin, R. (2023). Enhancing ProteinBERT: Integrating Intrinsically Disordered Proteins for Comprehensive Proteomic Predictions. 2023 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 3824–3831.
[10]. Vaswani, A. (2017). Attention is all you need. Advances in Neural Information Processing Systems.
[11]. Vig, J., Madani, A., Varshney, L. R., Xiong, C., Socher, R., & Rajani, N. F. (2021). BERTology Meets Biology: Interpreting Attention in Protein Language Models (arXiv:2006.15222).
[12]. Morozov, V., Rodrigues, C. H., & Ascher, D. B. (2023). CSM-Toxin: a web-server for predicting protein toxicity. Pharmaceutics, 15(2), 431.
Cite this article
Li,X. (2024). ProteinBERT Algorithms: Applications in Antimicrobial Peptides Classification, Intrinsically Disordered Protein Prediction, and Toxicity Analysis. Theoretical and Natural Science,71,99-107.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of ICBioMed 2024 Workshop: Computational Proteomics in Drug Discovery and Development from Medicinal Plants
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Brandes, N., Ofer, D., Peleg, Y., Rappoport, N., & Linial, M. (2022). ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics, 38(8), 2102–2110.
[2]. Gene Ontology Consortium. (2004). The Gene Ontology (GO) database and informatics resource. Nucleic acids research, 32(suppl_1), D258-D261.
[3]. Oldfield, C. J., & Dunker, A. K. (2014). Intrinsically disordered proteins and intrinsically disordered protein regions. Annual review of biochemistry, 83(1), 553-584.
[4]. Jain, A., & Kihara, D. (2019). NNTox: Gene Ontology-Based Protein Toxicity Prediction Using Neural Network. Scientific Reports, 9(1), 17923.
[5]. Lu, J., Zhang, H., Jin, C., & Quan, X. (2023). A Novel Classification Method for Antimicrobial Peptides Based on ProteinBERT. 2023 42nd Chinese Control Conference (CCC), 8437–8442.
[6]. Brandes, N., Ofer, D., Peleg, Y., Rappoport, N., & Linial, M. (2022). ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics, 38(8), 2102-2110.
[7]. Zhu, X., Guo, C., Feng, H., Huang, Y., Feng, Y., Wang, X., & Wang, R. (2024). A Review of Key Technologies for Emotion Analysis Using Multimodal Information. Cognitive Computation, 1-27.
[8]. Wang R., Zhu J., Wang S., Wang T., Huang J., Zhu X. Multi-modal emotion recognition using tensor decomposition fusion and self-supervised multi-tasking. International Journal of Multimedia Information Retrieval, 2024, 13(4): 39.
[9]. Stone, J. D., Tabrizi, H. B., & Shamsuddin, R. (2023). Enhancing ProteinBERT: Integrating Intrinsically Disordered Proteins for Comprehensive Proteomic Predictions. 2023 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 3824–3831.
[10]. Vaswani, A. (2017). Attention is all you need. Advances in Neural Information Processing Systems.
[11]. Vig, J., Madani, A., Varshney, L. R., Xiong, C., Socher, R., & Rajani, N. F. (2021). BERTology Meets Biology: Interpreting Attention in Protein Language Models (arXiv:2006.15222).
[12]. Morozov, V., Rodrigues, C. H., & Ascher, D. B. (2023). CSM-Toxin: a web-server for predicting protein toxicity. Pharmaceutics, 15(2), 431.