







1. Introduction
1.1. The Pathogenesis of Parkinson’s Disease and Corresponding Treatments
Over time, extensive research literature has developed on the pathological alternations in the dopamine pathway in substantia nigra compacta (SNc), a structure located deep in the brain. Dopamine is a neurotransmitter that relays signals between connected dopaminergic neurons (dopamine-releasing cells). Dopaminergic neurons in SNc play an indispensable role in fine-turning movement, while they are fragile due to their extensive branches and high energy demand for sending signals [1]. Consequently, the reduction of available dopamine, attributed to degenerating dopaminergic neurons, was argued to be responsible for movement-related symptoms and confirmed by clinical investigation. Parkinson’s related symptoms develop when there is a 50-60% dopaminergic neuron loss and an 80-85% decrease in dopamine concentration in SNc. Though yet to be fully understood, the current literature proposes several possible cellular mechanisms for the loss of dopaminergic neurons: mitochondrial dysfunction, oxidative stress, and inflammation [2]. From a proteomic perspective, PD is characterized by the formation of the abnormal accumulation of proteins in brain neurons called Lewy bodies, whose major constituent is \( α \) -synuclein ( \( α \) -Syn). \( α \) -Syn damages lysosomes’ breakdown function, accelerating Lewy bodies’ development [3]. From a genetic perspective, some pathogenic variants are responsible for Parkinson’s disease. For instance, PRKN, PINK1, and PARK7 are genes related to mitochondrial dysfunction [4]; SNCA is a gene that facilitates the formation of \( α \) -Syn [5]; and gene MAPT facilitates the aggregation of tau, a protein that is also found in Alzheimer’s and frontotemporal dementia [6, 7].
Unfortunately, current treatments are symptomatic, focusing on alleviating symptoms without disease-changing ability [8]. The current medicine included dopamine-based ones for motor symptoms, selective serotonin reuptake inhibitors for psychiatric disorders, and cholinesterase inhibitors for cognitive issues. Among those medicines, levodopa (L-DOPA), an amino acid that can pass through the blood-brain barrier and supplement dopamine concentration, is widely used for clinical treatments and scientific research. Nevertheless, owing to their symptomatic nature and the degradation of neurons, drug tolerance will ultimately develop, and medication-resistant tremors or dyskinesias will aggravate [9]. Therefore, an essential issue in the literature is further exploring the unrevealed mechanism and searching for disease-modifying medicine.
Although previous research almost exclusively focuses on the degeneration of dopaminergic neurons in SNc [10, 11], other types of neurons are also involved: decrease of serotoninergic neurons in the brainstem and denervation in cardiac norepinephrinergic neurons [12]. Furthermore, the MRI analysis indicated that multiple brain regions were involved in the process of Parkinson’s disease. The MRI studies compared the volume differences as Parkinson’s disease progresses. According to Blair [13] and Pieperhoff [14], nearly every subcortical region shrinks along with the progression of Parkinson’s disease, including the hippocampus, amygdala, and Entorhinal Cortex, which could shed light on a better understanding of pathology and provide alternative treatments.
1.2. The Hippocampus and Pyramidal Cells
The hippocampus was suspected to be responsible for the dementia exhibited in Parkinson’s disease due to its imperative role in memory formation. The hippocampus, located in the deep region of the brain named the midbrain, is an indispensable part of spatial and temporal memory formation [15]. Previous experiments like the classical H.M. case showed that if the hippocampus region were damaged, the patient would likely fail to form new long-term memory [16], which is also one of the cognitive dysfunctions exhibited in Parkinson’s disease patients.
Many previous and current studies have shown that the hippocampus has PD-related lesions. From macroscopic observations, decreasing bilateral hippocampus volume was spotted in MRI observations of PD patients [13, 17, 18]. These volumetric changes have various microscopic explanations, including genes, mitochondria dysfunction, oxidative stress, Lewy bodies, and inflammation. Besides these, existing literature examined synaptic plasticity neurons to find out the underlying reasons behind the degeneration in the hippocampus. Costa et al. formulated that hippocampal long-term depression (LTP) is reduced in PD mice due to the deficits in NMDA receptors; L-DOPA administration can restore the abnormalities, suggesting that hippocampal dopaminergic pathways may contribute to PD-related cognitive dysfunction [10]. Moreover, genetic study elucidates the pathological changes accompanied by Parkinson’s disease. The mitochondrial dysfunction causing mutation in PARK2 (a gene that codes for parkin, a ubiquitin E3 ligase participating in mitochondrial homeostasis) was found to lead to the proliferation of excitatory hippocampal glutamate synapses, and consequently increasing synaptic vulnerability and eventually contributed to the loss of neurons [19]. On the other hand, other researchers cast doubts on the PD-induced hippocampus and occurrence by pointing out that no significant loss of hippocampal neurons was found, and reported discrepancies resulted from inaccurate measurements of neuron numbers [20, 21]. As a result, investigations in the hippocampus are promising for solving disputes and finding the solution for Parkinsonian cognitive impairment.
This research focuses on the pyramidal neurons, the primary neurons in the hippocampus (accounting for 70% of total neurons in the hippocampus). The hippocampus is composed of two major groups of neurons: projection neurons (granule cells and pyramidal cells) and interneurons ( with 16 divergent types [22]). Projection neurons are glutamatergic neurons (glutamate-releasing neurons) liberating excitatory signals, while interneurons are multifarious mediators forming inhibitory synapses (connections between neurons) with principal neurons. This paper specifically researches pyramidal neurons, characterized by their pyramidal or teardrop-shaped soma (neuron’s cell body) and multi-branched dendrites (the extension of neurons, which relay signals) (as shown in figure 1(a)). With manifold ion channels located throughout the neurons, pyramidal neurons set the foundation for cognitive functions like memorizing, learning, and thinking.
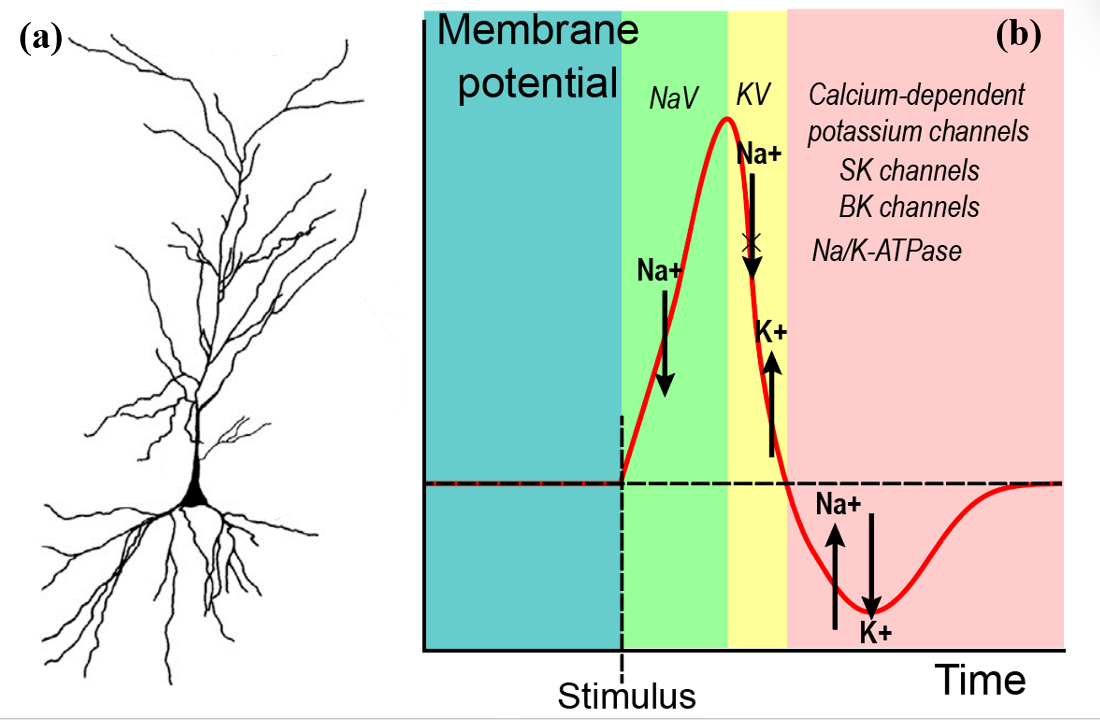
Figure 1. The schematics of pyramidal neurons and action potential and the related ion channels. (a) Camera tracings of representative pyramidal cells, showing cell structures. The triangular region is the soma, and the highly branched region is the dendrite that connects pyramidal neurons and others. Figure 1(a) is adapted from [23]. (b) Action potential is generated after the stimulus. There is an influx of sodium ions caused by the opening of NaV, causing the inner cellular potential to increase. Then, the NaV closes, and the outflow of potassium begins when KV opens. After that, multiple channels are involved to balance the membrane potential; some use energy, like N/K-ATPase, while others use facilitated diffusion.
1.3. Intrinsic Plasticity and Synaptic Plasticity
Plasticity, the basis of memory, is the manipulation of how neurons connect and react to stimuli by modulating the generation of action potential. Action potential starts from receiving neurotransmitters in their afferent synapse on the dendrite, a gap between neurons that converts received chemical signals to electrical voltage and passes the voltage to soma. Voltages are summed. If the summed voltage exceeds the critical voltage, the axon hillock will initiate the new action potential (As shown in figure 1(b)). Whereafter, the signals travel through the axon to synapses, where neurotransmitters are released and received by succeeding neurons. The signal transduction relays the influx and outflow of sodium (Na+) and potassium (K+) ions. The influx of sodium is regulated by voltage-gated sodium channels (NaV); the leakage of potassium is controlled by voltage-gated potassium channels (KV). The return of resting potential is adjusted by calcium-dependent potassium channels (which include two subtypes: small conductance calcium-activated potassium channels, or SK channels, and large conductance calcium-activated potassium channels, or BK channels), and sodium-potassium pumps (Na/K-ATPase).
Intrinsic plasticity and synaptic plasticity are two mechanisms that are involved in the process of action potential generation. Intrinsic plasticity is defined as the change in ion channel activity, number, and distribution [24] that affects neurons’ fundamental mechanisms, including synaptic integration, subthreshold propagation, and spike generation [25]. Intrinsic plasticity occurs in axons, dendrites, and cell bodies. On the other hand, synaptic plasticity refers to changes in the strength of synaptic connections based on external stimuli, or specifically, the number and type of ion receptors, ion channels, and the membrane electrical potential. There are two key components: long-term depression (LTD), and long-term potentiation (LTP), corresponding to the decrease and increase of membrane potential, caused by the widely researched NMDA and AMPA receptors [26]. Since the neuron’s electrical potential must exceed the threshold to fire, LTD and LTP affect the difficulty of firing. While both types of plasticity include changes in ion channels: calcium-related, voltage-gated, and ligand-gated ion channels, the differences between the two types of plasticity included the location and types of ion channels. Together, intrinsic plasticity and synaptic plasticity set the foundation for memory.
There are emerging research interests in intrinsic plasticity, since it can explain why animals are capable of learning after one stimulus, while synaptic plasticity requires multiple stimuli to have an effect. Besides, synaptic plasticity modulates the transduction of neuro signals more extensively because intrinsic plasticity determines the possibility that action potential is initiated in the axon hillock, as well as the shape of action potential and spike sequence (such as frequency and spike number). There is considerable research about intrinsic plasticity that enhances the understanding of diseases, including epilepsy, chronic pain, and addiction [27–29]. Some researchers found that the K+ channel is downregulated in epilepsy patients [30, 31], and knocking out K+ channels (Kv4.2) increases hyperexcitability in mice [32].
1.4. Statement of Purpose
This paper investigates the intrinsic plasticity and ion channels malfunctioning in Parkinson’s disease mice by calculating intrinsic factors. As the first attempt to investigate this area, it could shed light on a better understanding of Parkinson’s disease mechanism and help find potential pharmaceutical targets.
2. Materials and Methods
2.1. Animal model
The Parkinson’s disease mice were prepared by genetic modification using the MitoPark model proposed [33]. This model disrupts mitochondrial function in dopaminergic neurons by knocking out mitochondrial transcription factor A (mftA) [34]. As a pivotal protein in mitochondrial DNA expression and maintenance, mftA knockout causes mitochondrial damage in mice. These mice display progressive dopaminergic degeneration, motor impairments, and varying responses in L-DOPA treatment. In this paper, the “PD” group represents the genetically modified mice, while the “Intact” group represents not manipulated ones that serve as a control.
2.2. Slice preparation, pharmacological manipulations, and whole-cell recording
The neuron samples were produced by making brain slices of the hippocampus region in the mice. Multiple drugs that block synaptic connections (by blocking receptors and ion channels on the synapses) were used to fix synaptic plasticity and measure intrinsic plasticity. The data were acquired from the Margoliash Lab at the University of Chicago using the whole-cell current patch clamp. The cell received 100 \( pA \) currents, and the cross-membrane voltage was measured by the patch clamp, sampled at 10 \( kHz \) for both cell types. Parkinson’s disease neurons (n=8) and intact neurons (n=10) are examined with a standardized procedure. The detailed methods are illustrated in the method section of this paper [35].
2.3. Pharmacological manipulation
A gravity perfusion system was used to apply pharmacological reagents. For blocking synaptic currents, antagonists of N-methyl-Daspartate (NMDA), AMPA/kainate, GABAA and GABAB receptors: 6-cyano-7nitroquinoxaline-2,3-dione (CNQX, 10 μM; no. 0190, Tocris), (RS)-3-(2Carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP, 10 μM; no. 0173, Tocris), (3-Aminopropyl)(diethoxymethyl) phosphinic acid (CGP 35348, 50 μM; no. 123690-79-9, Santa Cruz) were used. SR 95531 Hydrobromide (Gabazine, 10 μM; no. 104104-50-9, Santa Cruz) was used to block GABAA receptors. DMSO was the solvent for CNQX and PTX (final concentration 0.1%), while water was the solvent for CPP, CGP 35348 ,and Gabazine. In current clamp experiments, in addition to synaptic antagonists, the channel antagonists were added: cadmium chloride (300 μM; no. 10108-64-2, Sigma), tetraethylammonium chloride (TEA, 25 mM; no. T2265, Sigma), cesium chloride (5 mM; no. 4739, Tocris), and tetrodotoxin (TTX, 5 μM; no. BML-NA120-0001, Enzo). Throughout the trials, some neurons were lost because of the numerous harmful medications that were applied to the slice. Only those that remained healthy throughout all drug treatments are reported.
2.4. Spike features analysis
To compare the change of intrinsic plasticity in hippocampal pyramidal neurons, several features were selected based on the standard choice from the literature [28]. Figure 2(a) is the recording of the current patch clamp, which is called a spike train because it has several spikes. Each spike in the spike train is called action potential (AP), which has three key components: threshold, peak, and afterhyperpolarization. Threshold (Thr) is defined as the voltage where the instantaneous rate of change for the neuron electrical potential is greater than 10 \( mV/0.1ms \) , and the instantaneous rate of change is defined as \( \frac{dV}{dt}(t)=\frac{{V_{i+1}}-{V_{i}}}{{t_{i+1}}-{t_{1}}} \) . Peak is the highest voltage within an action potential. Afterhyperpolarization (AHP) is the minimum voltage after the peak. Several other features, including amplitude (the voltage difference between peak and threshold), time to peak-peak (TTP-peak) (the time interval between threshold to peak), time to peak-after hyperpolarization (TTP-AHP) (the time interval between peak and AHP). Other features include spike width (the time interval between the AP’s time to reach ascending half-amplitude and descending half-amplitude), spike count (the number of AP in the spike train), spike attenuation (the decrease of the peak from the first AP to the last AP), and first spike latency (Time between current injection and the AP reach the peak).
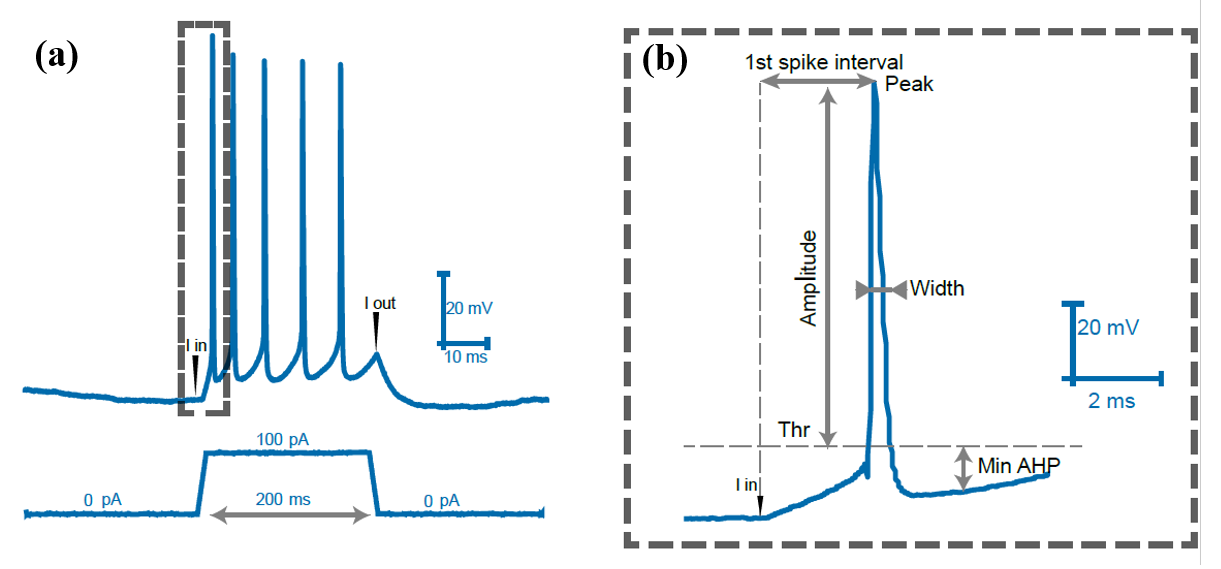
Figure 2. The recording from the current clamp and the schematic diagram of the features extracted. (a) This figure is an example of a whole-cell current patch clamp recording spike train acquired when a current of 100 \( pA \) was injected into a pyramidal neuron for 200 \( ms \) . The graph above records membrane voltage recording; below is the corresponding current injection. The “I in” and “I out” in this figure represent where the injection and removal of electrical currents correspond to the voltage. (b) figure 2(b) is an extraction of one action potential (AP) from figure 2(a). It shows the features that are used in the following analysis. The threshold (Thr), amplitude, peak, width, and minimum afterhyperpolarization (min AHP) are annotated on the graph.
3. Results
3.1. PD pyramidal neurons exhibit higher frequency and more morphological changes.
PD and intact (the control group) neuro-recordings are compared to unravel changes in intrinsic plasticity; the results are shown in Figure 3. The similarity between the two sets of data is that pyramidal neurons continue to fire after inputting the same amount of current, thus triggering multiple action potentials. In addition, within each group, the first spike has the highest peak voltage, and later spikes have a lower peak voltage than the first spike.
On the other hand, the spike train differs in some aspects between the PD and intact cases. First, the most apparent change is the increase in firing frequency. PD cases had significantly more spikes and shorter inter-spike intervals during the same stimulation period. Previous research suggested that this could be due to the downregulation of voltage-activated potassium channels [36]. Second, when spike waveforms of each spike were extracted and superimposed, they demonstrated a variety of waveforms in PD cases. These differences suggest the PD pathology and set the foundation for the subsequent quantitative analysis based on features calculated to evaluate differences in intrinsic plasticity systematically.
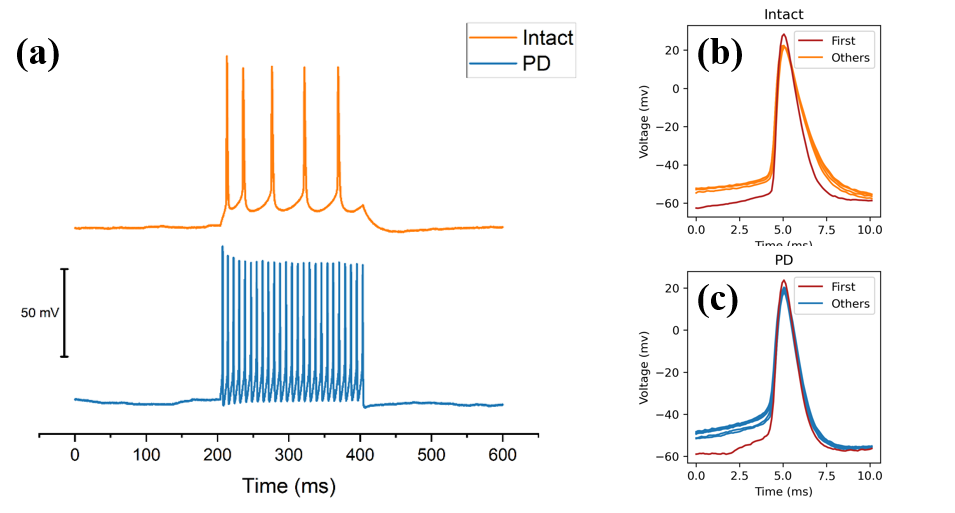
Figure 3. Spike train from PD and intact mice, and the morphological comparison between each spike. (a) An example of spike train data gathered from hippocampal pyramidal neurons from PD mice and intact mice, each of them was acquired from a current patch clamp with the 100 \( pA \) current stimulation for 200 milliseconds. The blue line is PD pyramidal neurons, and the orange line is from intact neurons. (b) The overlapping of action potential within one PD spike train. (c) The overlapping of action potential within one PD spike train. In figure 3(b) and figure 3 (c), the first spike is marked in red, and morphological differences can be seen in the graph.
3.2. Extracted Features Indicated Significant Differences Between PD and Intact.
A Mann-Whitney U test was performed to evaluate whether the intrinsic features of hippocampal neurons collected from animals exhibiting Parkinson’s disease differ from hippocampal neurons collected from intact (non-manipulated) animals. It can be seen from the data in figure 4 that there are apparent disparities in intrinsic plasticity features between PD and intact neurons, revealing the neurophysiology of PD pyramidal neurons in the hippocampus.
First, the PD cells have wide-ranging morphology. Strong evidence of decreasing peak voltage is found in this study (U = 12, p = .014), which also significantly reduced amplitude of AP (U = 17, p = .043). Sodium channels are principally responsible for controlling peak. Therefore, it is likely that the decrease in peak is a result of the downregulation of the sodium channel, which is supported by the calculation of the Nernst equation [37]. Furthermore, even though the TTP-peak (interval between reaching threshold and peak) is not significantly different between PD and intact group, a closer inspection of figure 4(a) shows that the boxplot of the TTP-peak of PD is much longer than that of intact, indicating that the TTP-peak of PD is more spread out than the TTP-peak of intact, which might be caused by the downregulation of voltage-gated sodium channels (NaV), and upregulation of the region responsible for inactivating NaV [38]. Additionally, the first spike latency is significantly decreased in the PD group, meaning PD neurons exhibit a higher level of excitability that takes significantly less time to react after stimulus.
Second, PD samples reveal abnormal changes in afterhyperpolarization. Figure 4(b) indicates a significant decrease (U = 14, p = .023) in TTP-AHP in PD neurons. TTP-AHP is a feature of the action potential controlled primarily by calcium-dependent potassium channels (including SK and BK channels). While we did not test this pharmacologically, the increase in TTP-AHP in PD neurons hints clearly to an upregulation in the calcium-dependent potassium channels. This finding broadly supports the work of other researchers linking BK channels to learning [39] and SK channels with locus coeruleus in PD cases [40].
Surprisingly, not all features extracted exhibit significant differences. There are no significant differences in features of width and threshold (as shown in figure 4(a) and figure 4(b), indicating that the potassium currents (controlling the width and threshold) are not significantly altered.
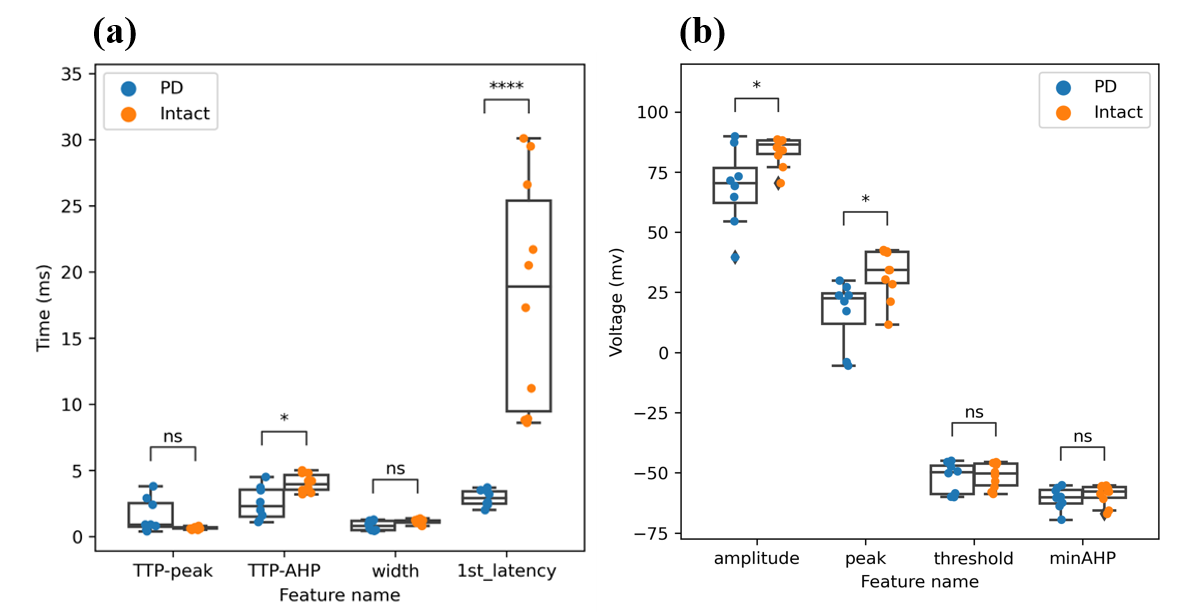
Figure 4. There are evident differences in firing patterns in the first spike among PD and intact mice. By analyzing features extracted from the first AP of each electrical stimulation, a significant increase in PD mice of TTP-AHP, peak, and amplitude is discovered among PD and intact mice. (a)The features in micro-seconds include TTP-peak (time to peak-peak), TTP-AHP (time-to-peak-afterhyperpolarization), and first spike latency. (b)The microvolt features include amplitude, peak, threshold, and minAHP (minimum afterhyperpolarization).
4. Discussion
This study’s data support that the intrinsic plasticity of hippocampal pyramidal neurons is altered in the PD case, especially in spike count (spike frequency), first spike latency, peak, and TTP-AHP. This study was the first to investigate intrinsic plasticity in hippocampal neurons using quantifiable features. These features could shed light on the preventive treatment of Parkinson’s disease.
The observed augmentation in spike count and initial spike latency signifies that under the influence of electrical currents, neurons in PD exhibit a heightened propensity to initiate action potentials. This increased excitability is consistent with prior studies indicating excitotoxicity in PD [41], suggesting that hippocampal pyramidal neurons exhibit excitotoxicity in both SNc and hippocampus, supporting the MRI observation of hippocampal atrophy [13, 14].
Over-excitability is posited to cause an influx of calcium ions, damaging mitochondria, and increasing the likelihood of mitochondrial dysfunction. This, in turn, can lead to cell death and contribute to neurodegenerative diseases such as Alzheimer’s and Parkinson’s [42]. Earlier studies have ascribed this heightened excitability to dysfunctional NMDA receptors [43]. However, the findings of this research are unexpected, given that all synaptic receptors were inhibited during neuronal treatment. This suggests that the hyperexcitability of neurons may be attributed to intrinsic ion channels not previously identified in the literature.
The observed reduction in peak voltages indicates premature closure of Nav channels following the initial stimulus, thereby allowing fewer sodium ions to increase the intracellular potential. This phenomenon could alternatively be attributed to the premature opening of voltage-gated potassium channels (KV). Researchers already observed that NaV channels could lead to cellular death and cancer cell proliferation [44, 45].
This research also found a significant reduction of time to peak afterhyperpolarization, indicating that the SK and BK ion channels deactivated in time, and the neurons are more prepared for future firing. SK and BK channels were recently discovered correlating to cerebellar dysfunction and impaired mitochondria [46].
There are several possible uncertainties related to this finding. A note of caution is due to the relatively small sample size, and only a single level (100pA) of current was injected into the neurons. The small-scale setting in this work introduces more random errors in the experimental procedure and variation within neurons; 100pA current may not be representative enough for all possible stimulating conditions. Moreover, the hypothesis of ion channel malfunction lacks confirmation from pharmacological blockers of specific ion channels or genetic sequencing to check for upregulations or downregulation of these channels. Finally, the stochastic nature of neuron’s firing patterns could also lead to potential uncertainty about the findings in this research [47].
There are already some medicines that target the malfunctioning ion channels that could be applied to the suggested intrinsic plasticity channels, like the medicines that treat epilepsy and regulate the excessively activated sodium ion channels [48], chlorzoxazone that could alleviate the symptoms caused by SK and BK channels dysfunction [46]. It is possible that these medicines could be used as potential targets for Parkinson’s patients.
These alteration in intrinsic excitability suggests pathological functioning impairment in NaV, SK, and BK ion channels located in locations other than commonly believed synapses was present in PD model mice. Identifying malfunctioning ion channels in the hippocampus may provide pharmaceutical targets to slow down neurodegeneration and mitigate cognitive impairment.
Future investigations should concentrate on the functionality of ion channels in hippocampal pyramidal neurons. Optogenetic studies could be employed to manipulate specific neuronal channel activity, aiming to establish a direct link between ion channels and cognitive behaviors. It is anticipated that future therapies could address not only the symptoms of Parkinson’s disease but also its underlying cellular pathways.
5. Data and code availability
Python programming language was used to process the data. The graphs were generated using Matplotlib, Statannotations, and Adobe Illustrator. The data and code supporting this study’s findings are available upon reasonable request.
Acknowledgments
I would like to take this opportunity to express my sincere gratitude to Professor Daou in the Department of Organismal Biology & Anatomy, University of Chicago, for his invaluable guidance and support throughout this research project. This work was made possible thanks to his provision of the necessary data, and I am deeply appreciative of his contribution to this paper.
References
[1]. Mamelak M. Parkinson’s Disease, the Dopaminergic Neuron and Gammahydroxybutyrate. Neurol Ther 2018; 7: 5–11.
[2]. Wirdefeldt K, Adami H-O, Cole P, et al. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol 2011; 26: 1–58.
[3]. Hoffmann A-C, Minakaki G, Menges S, et al. Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci Rep 2019; 9: 544.
[4]. Correia Guedes L, Mestre T, Outeiro TF, et al. Are genetic and idiopathic forms of Parkinson’s disease the same disease? Journal of Neurochemistry 2020; 152: 515–522.
[5]. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. The Lancet Neurology 2019; 18: 1091–1102.
[6]. Bouchard M, Suchowersky O. Tauopathies: One Disease or Many? Canadian Journal of Neurological Sciences 2011; 38: 547–556.
[7]. Day JO, Mullin S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021; 12: 1006.
[8]. Armstrong MJ, Okun MS. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020; 323: 548–560.
[9]. Lane EL. L-DOPA for Parkinson’s disease—a bittersweet pill. European Journal of Neuroscience 2019; 49: 384–398.
[10]. Costa C, Sgobio C, Siliquini S, et al. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain 2012; 135: 1884–1899.
[11]. Fahn S, Jankovic J, Hallett M. Medical treatment of Parkinson disease. In: Principles and Practice of Movement Disorders. Elsevier, pp. 119–156.
[12]. Sawada H, Oeda T, Yamamoto K. Catecholamines and Neurodegeneration in Parkinson’s Disease—From Diagnostic Marker to Aggregations of α-Synuclein. Diagnostics (Basel) 2013; 3: 210–221.
[13]. Blair JC, Barrett MJ, Patrie J, et al. Brain MRI Reveals Ascending Atrophy in Parkinson’s Disease Across Severity. Frontiers in Neurology; 10, https://www.frontiersin.org/articles/10.3389/ fneur.2019.01329 (2019, accessed 24 July 2023).
[14]. Pieperhoff P, Südmeyer M, Dinkelbach L, et al. Regional changes of brain structure during progression of idiopathic Parkinson’s disease – A longitudinal study using deformation based morphometry. Cortex 2022; 151: 188–210.
[15]. Ryan L, Lin C-Y, Ketcham K, et al. The role of medial temporal lobe in retrieving spatial and nonspatial relations from episodic and semantic memory. Hippocampus 2010; 20: 11–18.
[16]. Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry 1957; 20: 11–21.
[17]. Říha P, Brabenec L, Mareček R, et al. The reduction of hippocampal volume in Parkinson’s disease. J Neural Transm (Vienna) 2022; 129: 575–580.
[18]. Summerfield C, Junqué C, Tolosa E, et al. Structural Brain Changes in Parkinson Disease With Dementia: A Voxel-Based Morphometry Study. Archives of Neurology 2005; 62: 281–285.
[19]. Helton TD, Otsuka T, Lee M-C, et al. Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proceedings of the National Academy of Sciences 2008; 105: 19492–19497.
[20]. Joelving F c., Billeskov R, Christensen J r., et al. Hippocampal neuron and glial cell numbers in Parkinson’s disease—A stereological study. Hippocampus 2006; 16: 826–833.
[21]. Korbo L, Amrein I, Lipp H-P, et al. No evidence for loss of hippocampal neurons in non-Alzheimer dementia patients. Acta Neurologica Scandinavica 2004; 109: 132–139.
[22]. Parra P, Gulyás AI, Miles R. How Many Subtypes of Inhibitory Cells in the Hippocampus? Neuron 1998; 20: 983–993.
[23]. Spencer RL, Bland ST. Chapter 5 - Hippocampus and Hippocampal Neurons∗. In: Fink G (ed) Stress: Physiology, Biochemistry, and Pathology. Academic Press, pp. 57–68.
[24]. Sehgal M, Song C, Ehlers VL, et al. Learning to learn - intrinsic plasticity as a metaplasticity mechanism for memory formation. Neurobiol Learn Mem 2013; 105: 186–199.
[25]. Daou A, Margoliash D. Intrinsic plasticity and birdsong learning. Neurobiology of Learning and Memory 2021; 180: 107407.
[26]. Pereyra M, Medina JH. AMPA Receptors: A Key Piece in the Puzzle of Memory Retrieval. Front Hum Neurosci 2021; 15: 729051.
[27]. Graef JD, Godwin DW. Intrinsic plasticity in acquired epilepsy: too much of a good thing? Neuroscientist 2010; 16: 487–495.
[28]. Kourrich S, Calu DJ, Bonci A. Intrinsic plasticity: an emerging player in addiction. Nat Rev Neurosci 2015; 16: 173–184.
[29]. Yang Z, Tan Q, Cheng D, et al. The Changes of Intrinsic Excitability of Pyramidal Neurons in Anterior Cingulate Cortex in Neuropathic Pain. Frontiers in Cellular Neuroscience; 12, https://www.frontiersin.org/articles/10.3389/fncel.2018.00436 (2018, accessed 9 July 2023).
[30]. Tsaur M-L, Sheng M, Lowenstein DH, et al. Differential expression of K+ channel mRNAs in the rat brain and down-regulation in the hippocampus following seizures. Neuron 1992; 8: 1055–1067.
[31]. Monaghan MM, Menegola M, Vacher H, et al. Altered expression and localization of hippocampal A-type potassium channel subunits in the pilocarpine-induced model of temporal lobe epilepsy. Neuroscience 2008; 156: 550–562.
[32]. Chen X, Yuan L-L, Zhao C, et al. Deletion of Kv4.2 Gene Eliminates Dendritic A-Type K+ Current and Enhances Induction of Long-Term Potentiation in Hippocampal CA1 Pyramidal Neurons. J Neurosci 2006; 26: 12143–12151.
[33]. Ekstrand MI, Galter D. The MitoPark Mouse - an animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat Disord 2009; 15 Suppl 3: S185-188.
[34]. Keane PC, Kurzawa M, Blain PG, et al. Mitochondrial Dysfunction in Parkinson’s Disease. Parkinson’s Disease 2011; 2011: e716871.
[35]. Daou A, Margoliash D. Intrinsic neuronal properties represent song and error in zebra finch vocal learning. Nat Commun 2020; 11: 952.
[36]. Lu T, Wade K, Hong H, et al. Ion channel mechanisms underlying frequency-firing patterns of the avian nucleus magnocellularis: A computational model. Channels (Austin) 2017; 11: 444–458.
[37]. Byrne JH. Ionic Mechanisms and Action Potentials (Section 1, Chapter 2) Neuroscience Online: An Electronic Textbook for the Neurosciences | Department of Neurobiology and Anatomy - The University of Texas Medical School at Houston, https://nba.uth.tmc.edu/neuroscience/ m/s1/chapter02.html (2023, accessed 1 August 2023).
[38]. Ahern CA. What activates inactivation? J Gen Physiol 2013; 142: 97–100.
[39]. Matthews EA, Disterhoft JF. Blocking the BK channel impedes acquisition of trace eyeblink conditioning. Learn Mem 2009; 16: 106–109.
[40]. Matschke LA, Komadowski MA, Stöhr A, et al. Enhanced firing of locus coeruleus neurons and SK channel dysfunction are conserved in distinct models of prodromal Parkinson’s disease. Sci Rep 2022; 12: 3180.
[41]. Ambrosi G, Cerri S, Blandini F. A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J Neural Transm 2014; 121: 849–859.
[42]. Verma M, Lizama BN, Chu CT. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Translational Neurodegeneration 2022; 11: 3.
[43]. Armada-Moreira A, Gomes JI, Pina CC, et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Frontiers in Cellular Neuroscience; 14, https://www.frontiersin.org/articles/10.3389/fncel.2020.00090 (2020, accessed 10 November 2023).
[44]. Bachmann M, Li W, Edwards MJ, et al. Voltage-Gated Potassium Channels as Regulators of Cell Death. Frontiers in Cell and Developmental Biology; 8, https://www.frontiersin.org/articles/ 10.3389/fcell.2020.611853 (2020, accessed 10 November 2023).
[45]. Serrano-Novillo C, Capera J, Colomer-Molera M, et al. Implication of Voltage-Gated Potassium Channels in Neoplastic Cell Proliferation. Cancers 2019; 11: 287.
[46]. Du X, Carvalho-de-Souza JL, Wei C, et al. Loss-of-function BK channel mutation causes impaired mitochondria and progressive cerebellar ataxia. Proc Natl Acad Sci USA 2020; 117: 6023–6034.
[47]. Mendonça PR, Vargas-Caballero M, Erdélyi F, et al. Stochastic and deterministic dynamics of intrinsically irregular firing in cortical inhibitory interneurons. eLife 2016; 5: e16475.
[48]. Waszkielewicz AM, Gunia A, Szkaradek N, et al. Ion channels as drug targets in central nervous system disorders. Curr Med Chem 2013; 20: 1241–1285.
Cite this article
Wu,A.Y. (2024). Changes in intrinsic excitability of hippocampal pyramidal cells in Parkinson’s disease model. Theoretical and Natural Science,32,23-33.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Mamelak M. Parkinson’s Disease, the Dopaminergic Neuron and Gammahydroxybutyrate. Neurol Ther 2018; 7: 5–11.
[2]. Wirdefeldt K, Adami H-O, Cole P, et al. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol 2011; 26: 1–58.
[3]. Hoffmann A-C, Minakaki G, Menges S, et al. Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci Rep 2019; 9: 544.
[4]. Correia Guedes L, Mestre T, Outeiro TF, et al. Are genetic and idiopathic forms of Parkinson’s disease the same disease? Journal of Neurochemistry 2020; 152: 515–522.
[5]. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. The Lancet Neurology 2019; 18: 1091–1102.
[6]. Bouchard M, Suchowersky O. Tauopathies: One Disease or Many? Canadian Journal of Neurological Sciences 2011; 38: 547–556.
[7]. Day JO, Mullin S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021; 12: 1006.
[8]. Armstrong MJ, Okun MS. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020; 323: 548–560.
[9]. Lane EL. L-DOPA for Parkinson’s disease—a bittersweet pill. European Journal of Neuroscience 2019; 49: 384–398.
[10]. Costa C, Sgobio C, Siliquini S, et al. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain 2012; 135: 1884–1899.
[11]. Fahn S, Jankovic J, Hallett M. Medical treatment of Parkinson disease. In: Principles and Practice of Movement Disorders. Elsevier, pp. 119–156.
[12]. Sawada H, Oeda T, Yamamoto K. Catecholamines and Neurodegeneration in Parkinson’s Disease—From Diagnostic Marker to Aggregations of α-Synuclein. Diagnostics (Basel) 2013; 3: 210–221.
[13]. Blair JC, Barrett MJ, Patrie J, et al. Brain MRI Reveals Ascending Atrophy in Parkinson’s Disease Across Severity. Frontiers in Neurology; 10, https://www.frontiersin.org/articles/10.3389/ fneur.2019.01329 (2019, accessed 24 July 2023).
[14]. Pieperhoff P, Südmeyer M, Dinkelbach L, et al. Regional changes of brain structure during progression of idiopathic Parkinson’s disease – A longitudinal study using deformation based morphometry. Cortex 2022; 151: 188–210.
[15]. Ryan L, Lin C-Y, Ketcham K, et al. The role of medial temporal lobe in retrieving spatial and nonspatial relations from episodic and semantic memory. Hippocampus 2010; 20: 11–18.
[16]. Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry 1957; 20: 11–21.
[17]. Říha P, Brabenec L, Mareček R, et al. The reduction of hippocampal volume in Parkinson’s disease. J Neural Transm (Vienna) 2022; 129: 575–580.
[18]. Summerfield C, Junqué C, Tolosa E, et al. Structural Brain Changes in Parkinson Disease With Dementia: A Voxel-Based Morphometry Study. Archives of Neurology 2005; 62: 281–285.
[19]. Helton TD, Otsuka T, Lee M-C, et al. Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proceedings of the National Academy of Sciences 2008; 105: 19492–19497.
[20]. Joelving F c., Billeskov R, Christensen J r., et al. Hippocampal neuron and glial cell numbers in Parkinson’s disease—A stereological study. Hippocampus 2006; 16: 826–833.
[21]. Korbo L, Amrein I, Lipp H-P, et al. No evidence for loss of hippocampal neurons in non-Alzheimer dementia patients. Acta Neurologica Scandinavica 2004; 109: 132–139.
[22]. Parra P, Gulyás AI, Miles R. How Many Subtypes of Inhibitory Cells in the Hippocampus? Neuron 1998; 20: 983–993.
[23]. Spencer RL, Bland ST. Chapter 5 - Hippocampus and Hippocampal Neurons∗. In: Fink G (ed) Stress: Physiology, Biochemistry, and Pathology. Academic Press, pp. 57–68.
[24]. Sehgal M, Song C, Ehlers VL, et al. Learning to learn - intrinsic plasticity as a metaplasticity mechanism for memory formation. Neurobiol Learn Mem 2013; 105: 186–199.
[25]. Daou A, Margoliash D. Intrinsic plasticity and birdsong learning. Neurobiology of Learning and Memory 2021; 180: 107407.
[26]. Pereyra M, Medina JH. AMPA Receptors: A Key Piece in the Puzzle of Memory Retrieval. Front Hum Neurosci 2021; 15: 729051.
[27]. Graef JD, Godwin DW. Intrinsic plasticity in acquired epilepsy: too much of a good thing? Neuroscientist 2010; 16: 487–495.
[28]. Kourrich S, Calu DJ, Bonci A. Intrinsic plasticity: an emerging player in addiction. Nat Rev Neurosci 2015; 16: 173–184.
[29]. Yang Z, Tan Q, Cheng D, et al. The Changes of Intrinsic Excitability of Pyramidal Neurons in Anterior Cingulate Cortex in Neuropathic Pain. Frontiers in Cellular Neuroscience; 12, https://www.frontiersin.org/articles/10.3389/fncel.2018.00436 (2018, accessed 9 July 2023).
[30]. Tsaur M-L, Sheng M, Lowenstein DH, et al. Differential expression of K+ channel mRNAs in the rat brain and down-regulation in the hippocampus following seizures. Neuron 1992; 8: 1055–1067.
[31]. Monaghan MM, Menegola M, Vacher H, et al. Altered expression and localization of hippocampal A-type potassium channel subunits in the pilocarpine-induced model of temporal lobe epilepsy. Neuroscience 2008; 156: 550–562.
[32]. Chen X, Yuan L-L, Zhao C, et al. Deletion of Kv4.2 Gene Eliminates Dendritic A-Type K+ Current and Enhances Induction of Long-Term Potentiation in Hippocampal CA1 Pyramidal Neurons. J Neurosci 2006; 26: 12143–12151.
[33]. Ekstrand MI, Galter D. The MitoPark Mouse - an animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat Disord 2009; 15 Suppl 3: S185-188.
[34]. Keane PC, Kurzawa M, Blain PG, et al. Mitochondrial Dysfunction in Parkinson’s Disease. Parkinson’s Disease 2011; 2011: e716871.
[35]. Daou A, Margoliash D. Intrinsic neuronal properties represent song and error in zebra finch vocal learning. Nat Commun 2020; 11: 952.
[36]. Lu T, Wade K, Hong H, et al. Ion channel mechanisms underlying frequency-firing patterns of the avian nucleus magnocellularis: A computational model. Channels (Austin) 2017; 11: 444–458.
[37]. Byrne JH. Ionic Mechanisms and Action Potentials (Section 1, Chapter 2) Neuroscience Online: An Electronic Textbook for the Neurosciences | Department of Neurobiology and Anatomy - The University of Texas Medical School at Houston, https://nba.uth.tmc.edu/neuroscience/ m/s1/chapter02.html (2023, accessed 1 August 2023).
[38]. Ahern CA. What activates inactivation? J Gen Physiol 2013; 142: 97–100.
[39]. Matthews EA, Disterhoft JF. Blocking the BK channel impedes acquisition of trace eyeblink conditioning. Learn Mem 2009; 16: 106–109.
[40]. Matschke LA, Komadowski MA, Stöhr A, et al. Enhanced firing of locus coeruleus neurons and SK channel dysfunction are conserved in distinct models of prodromal Parkinson’s disease. Sci Rep 2022; 12: 3180.
[41]. Ambrosi G, Cerri S, Blandini F. A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J Neural Transm 2014; 121: 849–859.
[42]. Verma M, Lizama BN, Chu CT. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Translational Neurodegeneration 2022; 11: 3.
[43]. Armada-Moreira A, Gomes JI, Pina CC, et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Frontiers in Cellular Neuroscience; 14, https://www.frontiersin.org/articles/10.3389/fncel.2020.00090 (2020, accessed 10 November 2023).
[44]. Bachmann M, Li W, Edwards MJ, et al. Voltage-Gated Potassium Channels as Regulators of Cell Death. Frontiers in Cell and Developmental Biology; 8, https://www.frontiersin.org/articles/ 10.3389/fcell.2020.611853 (2020, accessed 10 November 2023).
[45]. Serrano-Novillo C, Capera J, Colomer-Molera M, et al. Implication of Voltage-Gated Potassium Channels in Neoplastic Cell Proliferation. Cancers 2019; 11: 287.
[46]. Du X, Carvalho-de-Souza JL, Wei C, et al. Loss-of-function BK channel mutation causes impaired mitochondria and progressive cerebellar ataxia. Proc Natl Acad Sci USA 2020; 117: 6023–6034.
[47]. Mendonça PR, Vargas-Caballero M, Erdélyi F, et al. Stochastic and deterministic dynamics of intrinsically irregular firing in cortical inhibitory interneurons. eLife 2016; 5: e16475.
[48]. Waszkielewicz AM, Gunia A, Szkaradek N, et al. Ion channels as drug targets in central nervous system disorders. Curr Med Chem 2013; 20: 1241–1285.