







1. Introduction
Psoriatic arthritis (PsA) is a common chronic inflammatory skin disease, which affects 1-3% of the world’s population. The PsA symptoms perform in heterogeneous musculoskeletal and non-musculoskeletal simultaneously. Erythema usually appears on the patient's skin and is covered with silvery white scales. The typical features are chronic, recurrent, benign papulosquamous lesions. The spontaneous remission rate of psoriatic arthritis is 40%. Nevertheless, it relapses easily. Traditional Chinese medicine treatment is of benefit to alleviate symptoms and promote the skin lesions to a certain context. The PsA mainly has two therapies, anterior uveitis and cool blood detoxification. Anterior uveitis therapy treat patients with cycloplegic agents, glucocorticoids, and non-steroidal anti-inflammatory eye drops. Arthropathic psoriasis acute stage is caused by rheumatic toxic heat, and the treatment should be mainly by cool blood detoxification, accompanied by heat clearing dehumidification and easing joint movements. The relief period is mostly caused by coldness and dampness obstruction or Yin deficiency of liver and kidney. Traditionally, patients should be treated by nourishing liver and kidney, warming meridian and dredging collaterals, which is very salutary to patients with psoriatic arthritis [1].
Nowadays, a more novel and highly effective drug, tofacitinib, has become a candidate for the treatment of the PsA. It can effectively treat PsA and a series of rheumatoid arthritis diseases, and its therapeutic mechanism is mainly through the inhibition of kinases to block the signal transduction of inflammatory cytokines. Kinases are enzymes that catalyze the transfer of phosphate groups from ATP to specific proteins or small biomolecules. When a molecule is phosphorylated, its activity, reactivity, and ability to bind to other molecules are greatly increased. Kinases play an indispensable role in cell signal transduction, metabolism, protein regulation, etc. Nowadays, functional groups that can inhibit kinases are regarded as drug targets that can effectively treat a variety of diseases, and have been widely studied in the pharmacological and clinical fields for the development of modern drugs. Janus kinase 3 (JAK3), a hematopoietic cell-restricted tyrosine kinase, is an important target for drug synthesis. Dysregulation of Janus kinase and activator of transcription (JAK/STAT) is one of several pathogenic conditions leading to chronic inflammation. In the course of treating autoimmune diseases and cancer, the structure of compounds that can inhibit JAK has been deeply explored. To date, there are five JAK inhibitors that have been approved by the Food and Drug Administration (FDA) for the treatment of different diseases [2].
Tofacitinib, a molecular kinase inhibitor, is one of them, and its high-throughput screening and lead optimization of its role as a JAK3 inhibitor have led to its emergence as a treatment for autoimmune diseases and organ transplant rejection reaction. Tofacitinib could inhibit JAK3 effectively (IC50=1.6 nmol/L), and it also has some effect on the inhibition of JAK1, JAK2 and Tyk2. When cytokines bind to receptors on target cells, they can phosphorylate JAK and thus phosphorylate STAT, and then activate JAK-STAT signaling pathway to play a role in regulating transcription. Tofacitinib reduces the production of T cells and other inflammatory cells by inhibiting JAK-STAT signaling, thereby blocking the signaling of various inflammatory cytokines. Given that JAK kinase plays a critical role in cell growth regulation, innate and adaptive immunity, and that tofacitinib inhibits JAK kinase by preventing it from binding to extracellular sites of different receptors, thereby preventing the production of various inflammatory cytokines. Therefore, tofacitinib has a unique advantage in the treatment of inflammatory diseases. In addition to its effects on psoriatic arthritis, tofacitinib can also treat ulcerative colitis (UC). Further studies have demonstrated that tofacitinib can alleviate the symptoms of alopecia areata (AA) and COVID-19 patients and improve the level of physical signs of patients [3]. In 2012, tofacitinib was developed by Pfizer (CP-690550) and named Xeljanz, with the molecular formula C16H20N6O. Tofacitinib became the first JAK inhibitor to be approved for the treatment of rheumatoid arthritis [4]. Tofacitinib is less toxic, more effective and orally active than some of the existing immunosuppressants used in organ transplantation and autoimmune diseases. Tofacitinib has played a crucial part in many major modern diseases, including cancer, autoimmunity, inflammation, and cardiovascular diseases. As the first listed JAK selective inhibitors, however, consumers shrink back at the sight of its fancy price, 60 tablets/bottle cost up to $4284, on average, each piece cost up to $71. The high prices for ordinary family are clearly a huge burden. Fortunately, tofacitinib’s patent protection of synthesis preparation in China has expired in 2020, so we are to find out the most suitable synthetic route that have the lowest cost and comparatively high yield. The structure of tofacitinib can be divided into two parts: The (3R,4R)-disubstituted piperidine and the deaminated purine core, which can be pieced together by N-alkylation. The synthesis of (3R,4R)-disubstituted piperidine derivatives can be achieved from different commercially available starting materials, and piperidine is the main source of cost of the tofacitinib production, which is also the bottleneck of the process. Because its structure contains two chiral centers, it is necessary not only to solve the problem of enantioselectivity in preparation, but also to have a good diasteroselectivity, which undoubtedly brings difficulties to the final chiral resolution step.
Given tofacitinib’s outstanding performance in pharmaceutical field and its high cost, three synthetic routes of tofacitinib is analyzed by reverse transcription synthesis. The first synthetic route carries out the conversion of tartrate to tofacitinib citrate with comparatively low cost and high purity. The second synthetic route is the enantioselective total synthesis of tofacitinib, which was achieved by hydroxylation of 1-benzyl-4-methylpiperidin-3-ol with 4-piperidone as the starting material and proline as the catalyst. The third synthetic route use 4-methylpyridine-3-amine to prepare intermediate (3R,4R)-1-benzyl-N-4-dimethylpiperidine-3-amine, and then the mixture was resolved with p-xylene formyl tartrate to achieve a good chiral purity. Analysis of existing synthetic routes and comparison of their safety, convenience, cost and yield will be of great potential value for understanding drug efficacy and the development of antibody drugs based on this compound.
|
Figure 1. The chemical structure formula of the key intermediate [3]. |
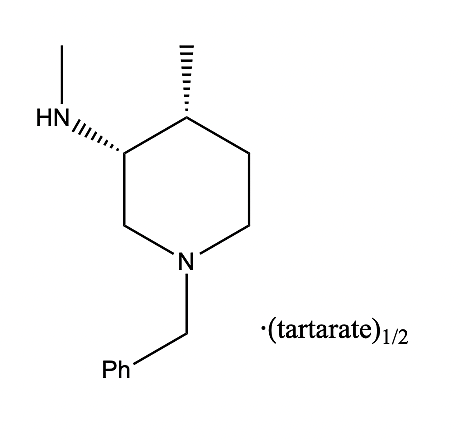
2. Synthetic Routes
Our first synthetic route uses an improved process to prepare tofacitinib citrate, which was approved by the FDA in November 2012 for the treatment of rheumatoid arthritis disease. A novel method was used to prepare (3R,4R)-(1-benzyl-4-methylpiperidine-3-methyl) amine salt, which is a key intermediate of tofacitinib citrate, and its chemical structure formula is shown in Fig. 1.
|
Figure 2. The traditional preparation method [3]. |
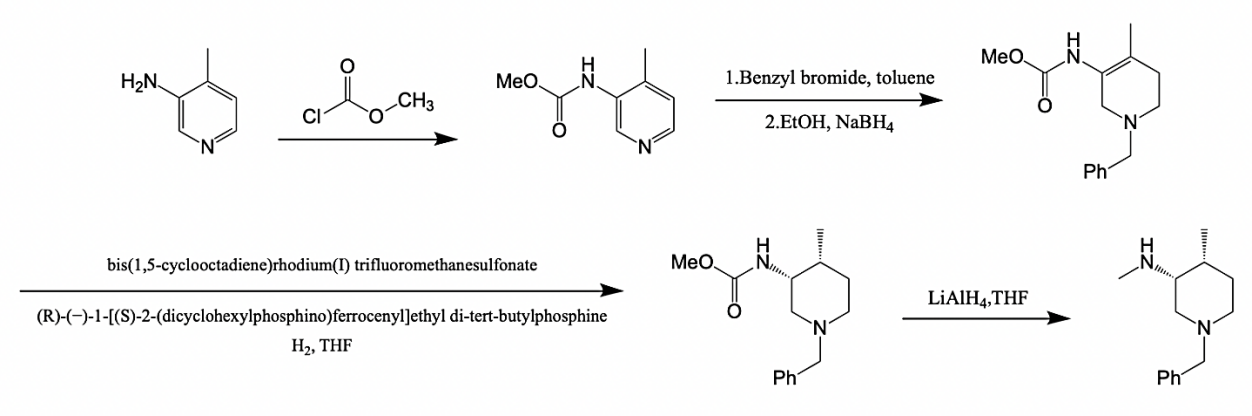
Fig. 2 shows a traditional way to compound (3R,4R)-(1-benzyl-4-methylpiperidine-3-methyl) amine salt. However, the product of the intermediate after hydrogenation reduction by tetrahydrofuran showed that its enantiomer overvolume (EE) was only 68%, and it contained 84% cis-isomers, which resulted in a poor chiral purity of the final product. Considering that the synthesis route is too cumbersome and requires expensive reagents such as bis (1, 5-cyclooctene) rhodium (I) trifluoromethane sulfonate, (R)(-)1-[(S)-2-(dicyclohexyl phosphine) ferrocenyl] ethylditert-butyl phosphine for catalytic reduction, Fig. 3 shows a cheaper and more convenient route for the synthesis of intermediate (3R,4R)-(1-benzyl-4-methylpiperidine-3-methyl) amine salt.
|
Figure 3. The new preparation method [3]. |
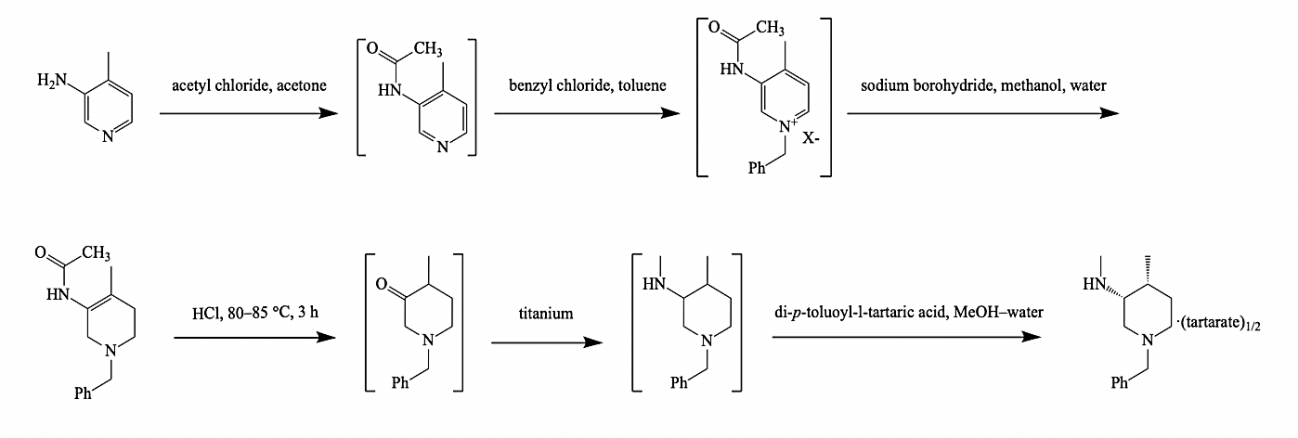
The synthesis route uses inexpensive and readily available 3-amino-4-methylpyridine as the starting material, put it in acetyl chloride and acetone solution for 8 h at room temperature to undergo n-acylation reaction to obtain amide intermediates, which has 95% yield. The pyridine ring of the amide intermediate occurs quaternary ammonium reaction by using benzyl chloride or benzyl bromide in toluene, followed by partial reduction with sodium borohydride in the mixture of methanol and water (1), with 91% yield. This route also optimizes the transition from compound (2) to compound (3) in acidic media. By making compound (2) react with two volumes of hydrochloric acid (36%) for 3 h at 65 °C, an enamine structure forms after hydrolysis. Because one of the two substituents on nitrogen is hydrogen, the enamine and imine formed are tautomers of each other. The enamine-imine tautomer is similar to the keto-enol tautomer, so the final intermediate is the tautomer of enamine, which is a ketone structure. Then, Borch reduction was carried out in the presence of titanium tetrisopropanol (IV), and the intermediate (3) was reduced and aminated with methanolic methylamine, and then we get compound (4) after the reductive amination reaction with sodium borohydride. It’s important to note that what's going on here is that the carbonyl group rather than oxime group is first added to methanolamine to get a secondary amine structure, which is then reduced by sodium borohydride (the catalyst here can also choose lithium aluminum tetrahydrogen or hydrogen nickel catalysis), because if secondary halogenated hydrocarbons are applied for amination, some of the products will be eliminated. Compound (4) was then resolved in a mixture of methanol and water with a resolution agent such as dibenzoyl-1-tartaric acid or xylene yl-1-tartaric acid to obtain the chiral intermediate (3R,4R)-(1-benzyl-4-methylpiperidine-3-methyl) amine salt, with a yield of 40%.
The condensation of (3R,4R)-(1-benzyl-4-methylpiperidine-3-methyl) amine salt and intermediate (6) is also a critical step in the formation of the backbone of tofacitinib citrate. Fig. 4 shows the reverse synthesis route of tofacitinib citrate. Tofacitinib citrate can be obtained by condensation of compound (9) with cyanoacetyl chloride or cyanoethyl acetate in non-nucleophilic bases such as DBU and n-butanol. The electropositive carbon attacks the lone pair electrons on nitrogen atoms, occurring nucleophilic addition reaction.
|
Figure 4. The flowchart of synthetic route 1 [3]. |
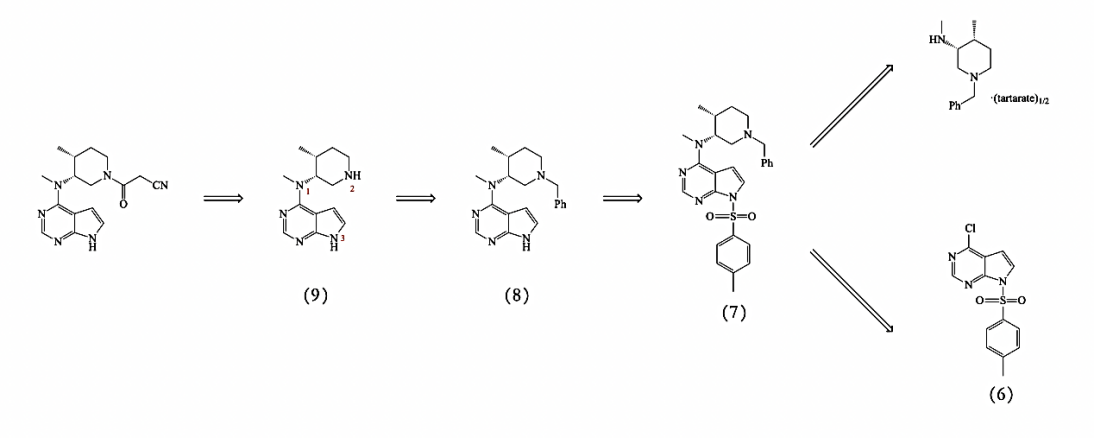
By breaking the single bond on No.1 nitrogen of compound (9) and we can readily observe that it can be split into two inferior compounds. However, there are two nitrogen atoms in compound (9) that we do not want them to participate in the reaction in the synthesis, so it is necessary to add protective groups to them in the previous reaction to prevent the occurrence of side reactions. Here, benzyl group was used to protect No.2 nitrogen and Tos was used to protect No.3 nitrogen. Compound (8) can be prepared by debenzylation with Pd/C, using water isopropyl alcohol and acetic acid as solvent. Hydrogenation reduction with palladium carbon as catalyst is a common method for debenzyl in organic synthesis. However, it should be noted that if the compound contains groups that are not stable for hydroreduction, the Pd/C method is no longer suitable, and the debenzylation reaction can be completed by using acyl chloride. Compound (7) can be prepared by coupling compound (6) and (3R,4R)-(1-benzyl-4-methylpiperidine-3-methyl) amine salts with excessive K2CO3. Compound (6) contains p-methyl benzenesulfonic acid base group, two oxygen atoms and the large π bond of the benzene ring can form a very stable resonance structure with the negative ions of the leaving group, so Tos is an ideal leaving group. Compound (7) can react with KOH, taking methanol as solventto, to remove toluene sulfonylate. The reaction material can be cooled and directly filtered to obtain a comparatively high purity compound (8) (96.3%) [3].
|
Figure 5. The inverse derivation of the synthesis route by the structure of tofacitinib [5]. |
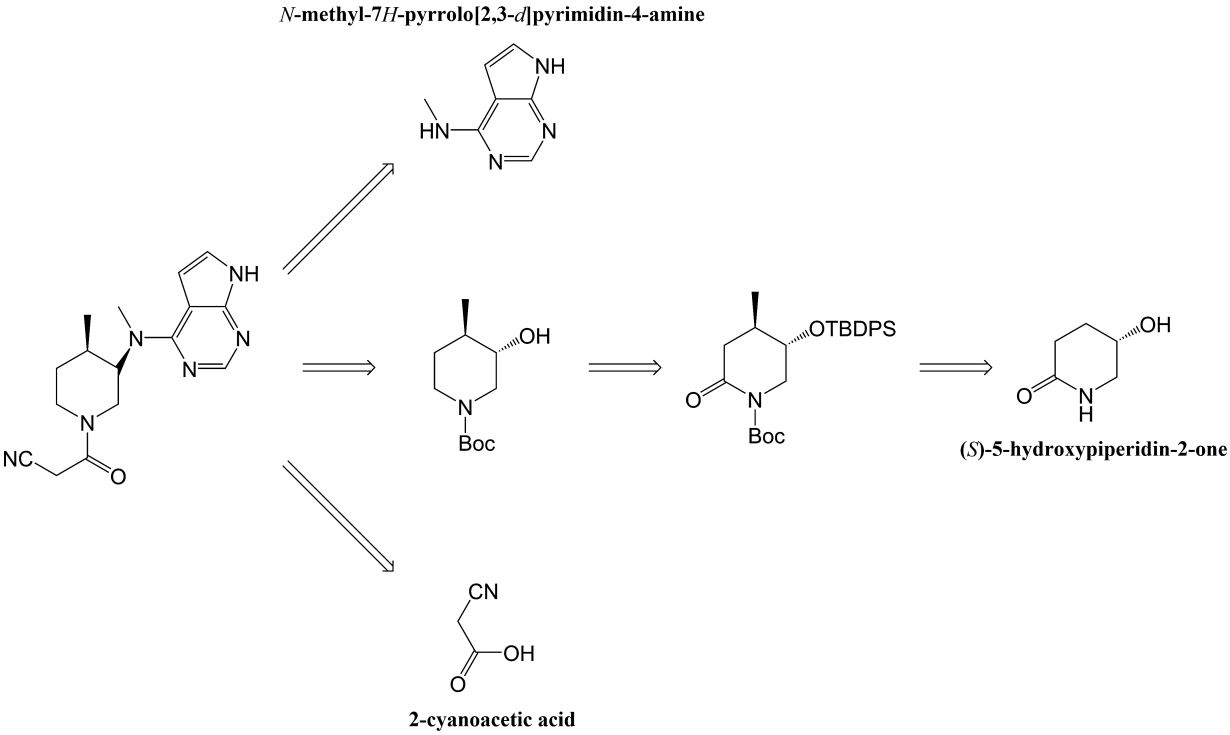
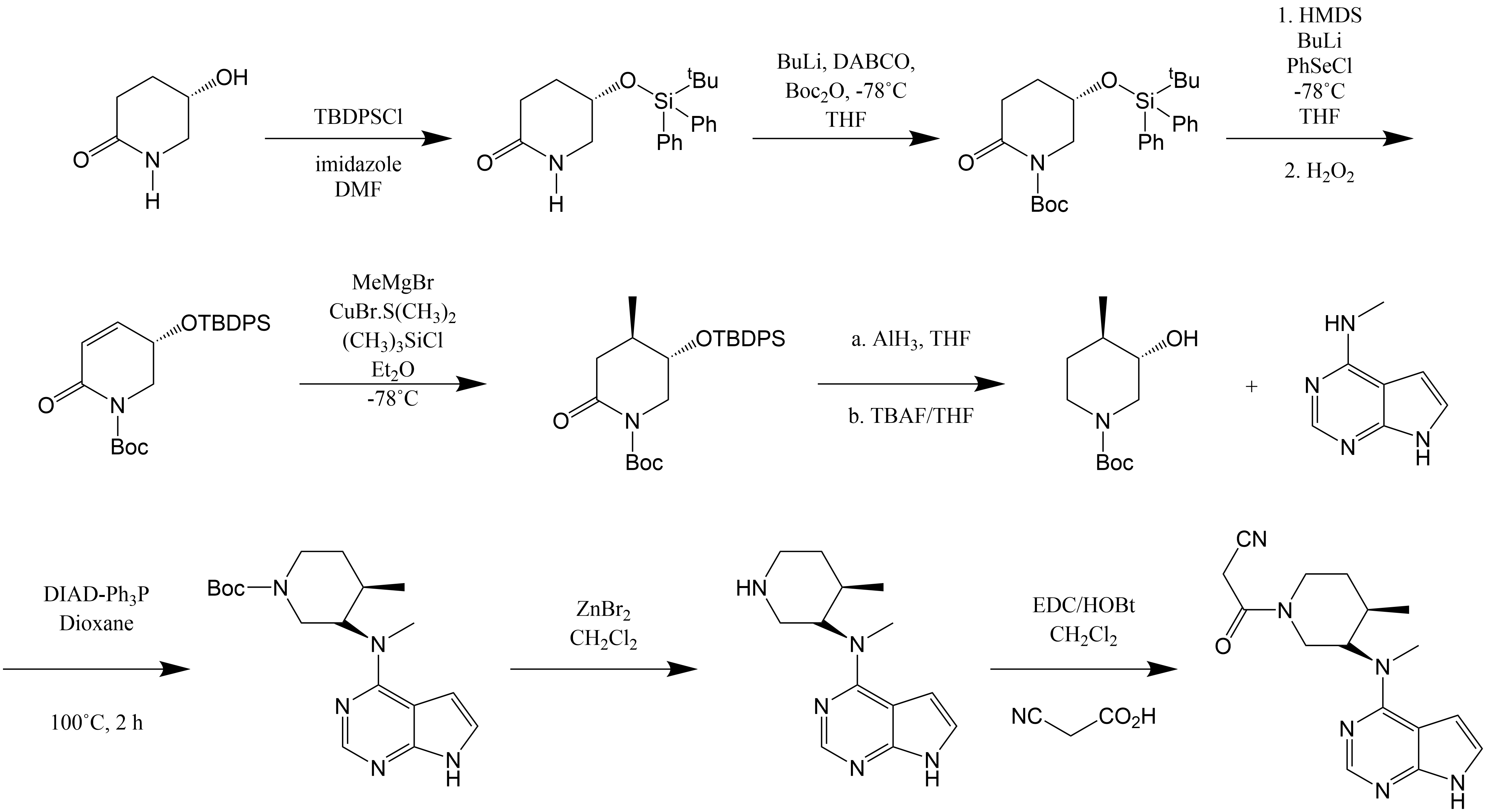
Fig. 5 is the inverse derivation of the synthesis route by the structure of tofacitinib, which can determine the main raw materials for the synthesis of tofacitinib. Here is how this synthetic route works. At first, hydroxyl group was protected with TBDPSCl/imidazole, where its yield was 96%. For the second reaction that it is to protect the amino group, the protecting group is butyloxycarboryl (Boc), and its yield was 85%. The third and fourth steps were to add the methyl group to the compound. As shown in Fig. 6, PhSeCl was added, and hydrogen peroxide was used for elimination reaction to form carbon-carbon double bond, and its yield was 60%. Then, for the following reaction, the a reaction was to remove the oxygen from the carbonyl group, and the b reaction is the removal of TBDPS protecting group, resulting in the formation of the key intermediate. The zinc bromide was used to remove the Boc protective group, and EDC/HOBt were used to catalyze its reaction with cyanoacetic acid. As a result, the final product was got.
|
Figure 6. The designed synthetic route [5]. |
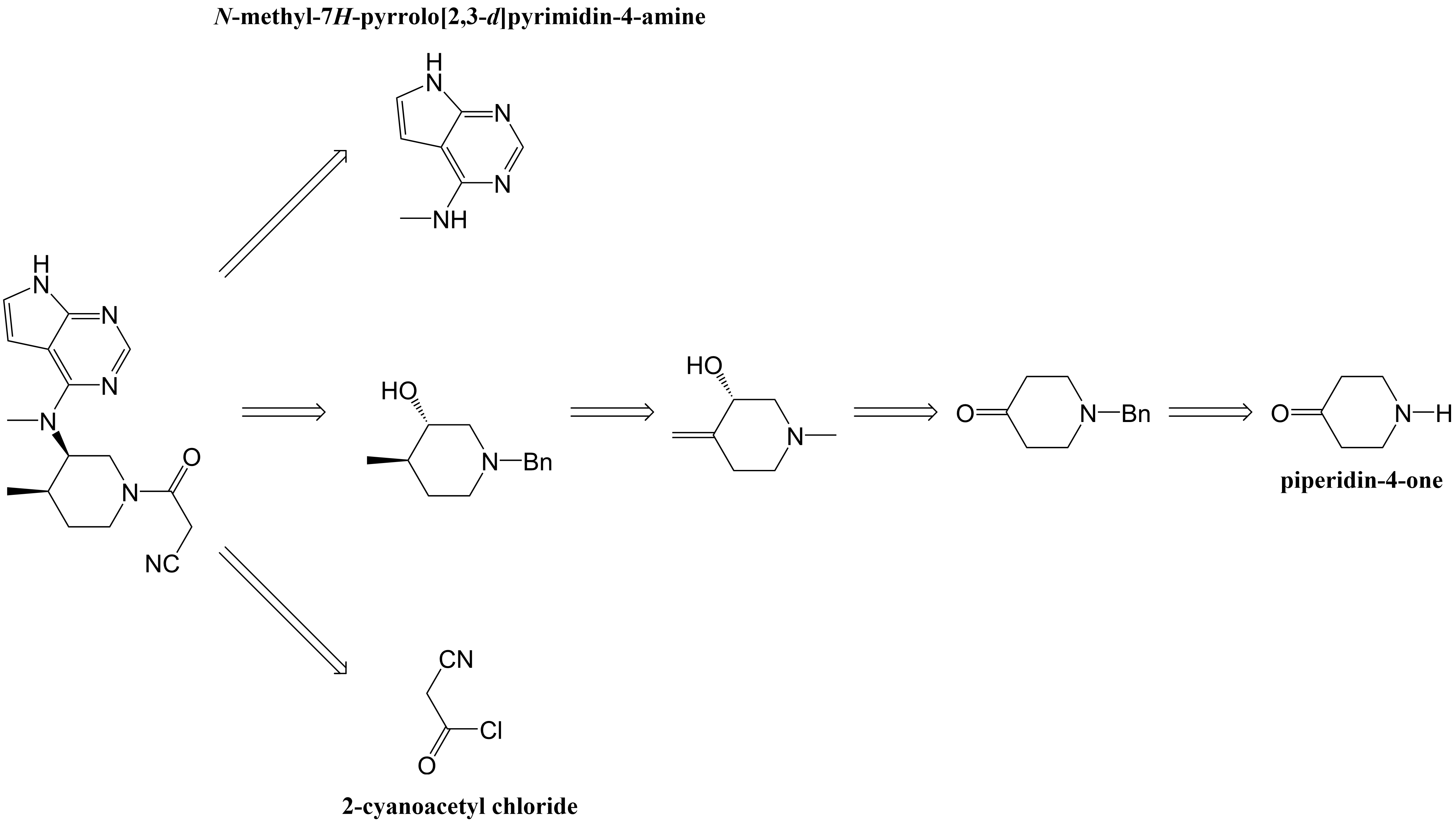
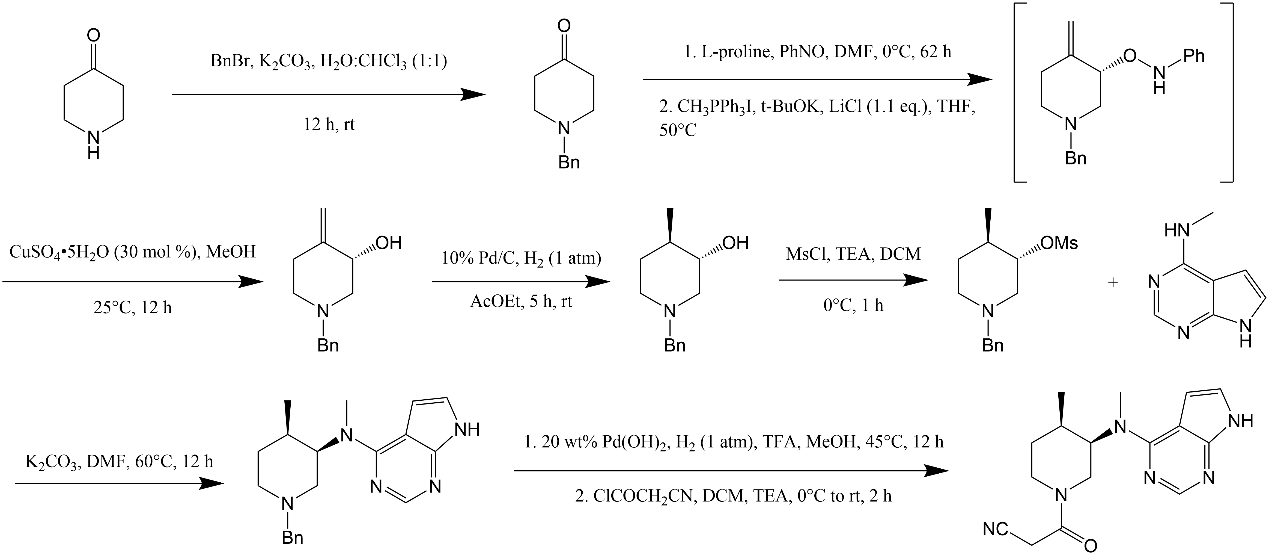
Then there’s the third synthetic route, it was similar to the second synthetic route, the synthetic route was deduced backwards by analyzing the structure of tofacitinib. As shown in Fig. 7, the first reaction is the formation of the protective group Bn (Benzyl) for nitrogen, and its yield was 87%. In the second reaction step, L-proline in the first step is an organic catalyst with chiral selectivity. In the second reaction, CH3PPh3I was used for witting reactions, t-BuOK was the base in the reaction and then the intermediate was formed. Copper sulfate pentahydrate was then added and the reaction continued for 12 hours to produce the corresponding chiral alcohols. The yield of these three steps was 47%. Although the yield here was low, its ee value was 97%. Then, at a reaction of 1 atm pressure of H2 and 10% Pd on carbon, it happened diastereoselective hydrogenation reaction, where its yield was 95% and its ee value was 96.8%. Then, MsCl and triethylamine was used for mesylation reaction. Next, the product and N-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine reacted with K2CO3 and DMF for 12 hours at 60 °C. Its yield was 81%. Finally, under 20 wt% Pd(OH)2 and 1 atm H2 pressure’s condition, they happened hydrogenation reaction. And then, it was combined with 2-cyanoacetyl chloride to make the final product (tofacitinib). For the synthetic route (Fig. 8), there were 8 reaction steps, the total yield was 22.4%, and the ee value was 96.8%.
|
Figure 7. The designed synthetic route [6]. |
For the first synthetic route, it focuses on optimizing the synthesis of an important intermediate for tofacitinib preparation and it’s (3R,4R)-(1-benzyl-4-methylpiperidin-3-yl)methylamine. The synthesis method was novel and it simplifies the tedious process of preparing the intermediate into a two-step process. The total yield was 15%, which was twice higher than that of the traditional method. That’s because the intermediate compound did not need to be separated. The synthesis route does not require the use of any expensive reagents and catalysts, and the cost is greatly reduced. But there are high temperature conditions, the intermediate needed to be heated to 110 °C when benzene methyl was added. Therefore, more attention needs to be paid to safety. The second synthetic route had more reaction steps and it was divided into 10 steps. Atomic economy (not considering recycling of by-products) was 66.80%. What’s more, some reaction conditions contained requirements of -78 °C, 100 °C, so more attention needs to be paid to safety. Next, the yield was low and the total yield was only 9.5%. Furthermore, the consumption was also large. But the ingredients are easier to gain. For the third synthetic route, the number of reaction steps was relatively fewer and it was divided into 8 steps, and the atomic economy (without considering the recycling of by-products) was 36.23%. The main reaction of this synthesis method was the olefin and hydrogenation reaction of Witting, and the key step was the chiral selection of organic catalyst (L-proline). The yield of the reaction was relatively high, the total yield was 22.4%. The temperature requirement of the reaction was not too high. The highest temperature was 60 °C, the lowest temperature was 0 °C, which is safe. But it used hydrogen, so it needs to take explosion protection measures. What’s more, some ingredients are difficult to obtain or they are very expensive.
|
Figure 8. Synthesis diagram of target substance [6]. |
3. Mechanism And Application
The therapeutic mechanism of JAK inhibitors is the inhibition of Janus (JAK) kinase. The JAKs subgroup consists of four members: JAK1, JAK2, JAK3 and Tyrosine kinase 2 (TYK2). The JAK1 and JAK3 kinases inhibited by Tofacitinib are involved in the signaling pathways of various cytokines involved in T cell development, activation and homeostasis. JAK1 plays an important role in the regulation of leukocyte and recombinant protein cytokine signaling as well as T-helper cell expansion, differentiation and memory formation, while JAK3 plays a major role in lymphoid development in the immune system.
In addition to the outstanding performance of tofacitinib in the treatment of psoriatic arthritis, studies have shown that JAK kinase inhibitors are also promising in the treatment of rheumatoid arthritis (RA), an immune-mediated disease through which cytokines transport intracellular signals. Tofacitinib, as a selective JAK inhibitor, can also inhibit the immune and inflammatory response of RA by blocking cellular inflammatory factors. Through control and parallel experiments, tofacitinib has a faster effect on the treatment of patients with poor effects of traditional biological reagents, and can significantly improve the symptoms and signs of patients, improve the physical function of patients, and the clinical benefit of tofacitinib is more than 24 months [7]. Tofacitinib also appears to have a better efficacy for COVID-19 pneumonia, the world’s mainstream infectious disease, with a lower risk of death or respiratory failure within 28 days in patients treated with tofacitinib compared with other biological agents [8].
In general, tofacitinib is a well-targeted therapy with high safety and fewer harmful side effects [9]. Compared with traditional drugs, tofacitinib has the advantage of oral administration. Therefore, tofacitinib is a good choice for patients who are intolerant to traditional anti-rheumatic drugs [10]. However, the effectiveness and safety of tofacitinib still need to be confirmed by more research data in the face of numerous individual physical differences in China's huge population base.
4. Conclusion
Tofacitinib, with its high-throughput screening and lead optimization of JAK3 inhibitors, is emerging as a treatment for autoimmune diseases and organ transplant rejection. In 2012, tofacitinib became the first JAK inhibitor approved for the treatment of rheumatoid arthritis. The pathogenesis of PsA is complex. Tofacitinib, as an inhibitor of JAK/STAT signal transduction pathway, may treat PsA in a variety of ways. However, there are few experiments on PsA model, and how the JAK-STAT pathway interacts with multiple pathways related to the pathogenesis of human psoriasis is still unclear. In synthesis, because of the enantiomers and the existence of enantiomers, need to purification of product, to cause a decline in yield again, the first synthetic route requires again purification, article 3 of the synthetic route chiral selection can be done, but the yield is still not very high, hope the future can be to choose a better synthetic route, increase the yield and purity of the drug.
Tofacitinib inhibits both Tyk2 and JAK kinases, however, JAK3 inhibition is more selective. For most not life-threatening tumor diseases, patients need long-term treatment, has the good isomer and kinase selective inhibitors can effectively reduce the side effects such as drug toxicity, so the development of preparation of selective kinase inhibitor can greatly reduce the occurrence of adverse events, improve the safety and efficacy of drugs. In addition, the design and synthesis of multi-target drugs is also of great interest, whose therapeutic mechanism is based on the regulation of multiple proteins rather than only acting on a single target. By identifying “master keys” that can recognize “multiple locks”, multi-target drugs can have synergistic effects in diseases such as cancer, inflammation, metabolic and neurological diseases, and achieve better treatment results. Multitarget drugs have higher efficacy, better safety, and fewer side effects than single-target drug combinations. The process of finding multi-target drugs also facilitates the repositioning of drugs that are already commercially available for the treatment of new diseases.
References
[1]. Stober C. (2021). Pathogenesis of psoriatic arthritis. Best practice & research. Clinical rheumatology, 35(2), 101694.
[2]. Xie, Z., Yang, X., Duan, Y., Han, J., & Liao, C. (2021). Small-Molecule Kinase Inhibitors for the Treatment of Nononcologic Diseases. Journal of medicinal chemistry, 64(3), 1283–1345.
[3]. Patil, Y.S., Bonde, N.L., Kekan, A.S., Sathe, D.G., & Das, A. (2014). An Improved and Efficient Process for the Preparation of Tofacitinib Citrate. Organic Process Research & Development, 18, 1714-1720.
[4]. Su, W., Chen, Z., Liu, M., et al. (2022). Design, synthesis and structure-activity relationship studies of pyrido[2,3-d] pyrimidin-7-ones as potent Janus Kinase 3 (JAK3) covalent inhibitors. Bioorganic & medicinal chemistry letters, 64, 128680.
[5]. Maricán, A., Simirgiotis, M. J., & Santos, L. S. (2013). Asymmetric total synthesis of Tofacitinib. Tetrahedron Letters, 54(37), 5096-5098.
[6]. Mane, K. D., Kamble, R. B., & Suryavanshi, G. (2021). Short enantioselective total synthesis of (+)-tofacitinib. Tetrahedron Letters, 67, 152838.
[7]. Sandborn, W. J., Su, C., Sands, B. E., et al. (2017). OCTAVE Induction 1, OCTAVE Induction 2, and OCTAVE Sustain Investigators. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med, 376(18), 1723-1736.
[8]. Guimarães, P. O., Quirk, D., Furtado, R. H., et al. (2021). Tofacitinib in Patients Hospitalized with Covid-19 Pneumonia. The New England journal of medicine, 385(5), 406–415.
[9]. Kim, D., Kobayashi, T., Voisin, B., et al. (2020). Targeted therapy guided by single-cell transcriptomic analysis in drug-induced hypersensitivity syndrome: a case report. Nature medicine, 26(2), 236–243.
[10]. Ruperto, N., Brunner, H. I., Synoverska, O., et al. (2021). Tofacitinib in juvenile idiopathic arthritis: a double-blind, placebo-controlled, withdrawal phase 3 randomised trial. Lancet (London, England), 398(10315), 1984–1996.
Cite this article
Sun,Y.;Zhu,H. (2023). Analysis and Comparison of Synthesis Route of Small Molecule Kinase Inhibitors Tofacitinib. Theoretical and Natural Science,4,220-228.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 2nd International Conference on Biological Engineering and Medical Science (ICBioMed 2022), Part II
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Stober C. (2021). Pathogenesis of psoriatic arthritis. Best practice & research. Clinical rheumatology, 35(2), 101694.
[2]. Xie, Z., Yang, X., Duan, Y., Han, J., & Liao, C. (2021). Small-Molecule Kinase Inhibitors for the Treatment of Nononcologic Diseases. Journal of medicinal chemistry, 64(3), 1283–1345.
[3]. Patil, Y.S., Bonde, N.L., Kekan, A.S., Sathe, D.G., & Das, A. (2014). An Improved and Efficient Process for the Preparation of Tofacitinib Citrate. Organic Process Research & Development, 18, 1714-1720.
[4]. Su, W., Chen, Z., Liu, M., et al. (2022). Design, synthesis and structure-activity relationship studies of pyrido[2,3-d] pyrimidin-7-ones as potent Janus Kinase 3 (JAK3) covalent inhibitors. Bioorganic & medicinal chemistry letters, 64, 128680.
[5]. Maricán, A., Simirgiotis, M. J., & Santos, L. S. (2013). Asymmetric total synthesis of Tofacitinib. Tetrahedron Letters, 54(37), 5096-5098.
[6]. Mane, K. D., Kamble, R. B., & Suryavanshi, G. (2021). Short enantioselective total synthesis of (+)-tofacitinib. Tetrahedron Letters, 67, 152838.
[7]. Sandborn, W. J., Su, C., Sands, B. E., et al. (2017). OCTAVE Induction 1, OCTAVE Induction 2, and OCTAVE Sustain Investigators. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med, 376(18), 1723-1736.
[8]. Guimarães, P. O., Quirk, D., Furtado, R. H., et al. (2021). Tofacitinib in Patients Hospitalized with Covid-19 Pneumonia. The New England journal of medicine, 385(5), 406–415.
[9]. Kim, D., Kobayashi, T., Voisin, B., et al. (2020). Targeted therapy guided by single-cell transcriptomic analysis in drug-induced hypersensitivity syndrome: a case report. Nature medicine, 26(2), 236–243.
[10]. Ruperto, N., Brunner, H. I., Synoverska, O., et al. (2021). Tofacitinib in juvenile idiopathic arthritis: a double-blind, placebo-controlled, withdrawal phase 3 randomised trial. Lancet (London, England), 398(10315), 1984–1996.