







1. Introduction
Proteins are the fundamental building blocks of life, playing crucial roles in virtually all biological processes. Despite significant advances in genome sequencing, protein structural and functional characterization, a substantial portion of the protein universe remains uncharted [1]. This "dark matter" of the protein universe consists of proteins whose structures or functions are still unknown. Elucidating the structure, function, and evolutionary relationships of these uncharacterized proteins is crucial for advancing our understanding of biology and for unlocking their potential in biotechnological and biomedical applications [2]. Traditionally, the study of uncharacterized proteins has relied on experimental methods such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy (cryo-EM) for structure determination. However, these techniques are labor-intensive, time-consuming, expensive, and often challenging to apply to proteins that are difficult to express, purify, or crystalize [3]. Computational approaches, such as homology-based modeling, have been used to predict protein structures based on sequence similarity to known structures. Nevertheless, these methods have limited applicability when studying proteins with low sequence homology (below 35% homology) to characterized proteins, which is often the case for the "dark matter" of the protein universe [4].
Recent breakthroughs in artificial intelligence (AI) have revolutionized the field of protein structure prediction. Deep learning-based methods, such as AlphaFold [5] and RoseTTAFold [6], have achieved unprecedented accuracy and speed in predicting protein structures from their amino acid sequences. These AI-enabled structure prediction methods have the potential to illuminate the "dark matter" of the protein universe by providing high-quality structural models for previously uncharacterized proteins [7]. By leveraging these predicted structures, researchers can gain valuable insights into the function, evolution, and potential design of new proteins for novel applications. This study seeks to highlight the recent AI-enabled advancements in the exploration of the "dark matter" of the protein universe through literature review and analyses. It focuses particularly on the breakthroughs in AI-driven prediction of protein and protein complex structures, the dramatic increase in accessible structural information, the improved annotation and understanding of previously uncharted protein families and functions, and AI-facilitated design of novel proteins. This review aims to enhance our understanding of AI's transformative impact on exploring and expanding the protein universe, and to inspire continued innovation and application in the field.
2. AI-enabled protein structure prediction methods and databases
The field of protein structure prediction has witnessed a remarkable transformation in recent years, largely driven by the advent of deep learning-based methods. These AI-enabled approaches have significantly outperformed traditional homology-based structure prediction methods and have set new benchmarks in speed and accuracy [1].
One of the most prominent breakthroughs in AI-based protein structure prediction is AlphaFold, developed by DeepMind [5, 8]. AlphaFold employs a deep learning model that leverages evolutionary, physical, and geometric constraints to predict the 3D structure of a protein from its amino acid sequence. The model was trained on a vast dataset of experimentally determined protein structures and has demonstrated exceptional performance in the Critical Assessment of Protein Structure Prediction (CASP) competition [9]. AlphaFold's success has been attributed to its ability to capture complex patterns and long-range interactions between amino acid residues, enabling it to generate highly accurate structural models even for proteins with limited or no sequence homology to known structures.
Another notable AI-based method is RoseTTAFold, developed by the Baker Lab at the University of Washington [6]. RoseTTAFold combines deep learning with a three-track neural network architecture to predict protein structures. The method incorporates co-evolutionary information, protein sequence, and residue-residue distances, and atom coordinates to generate high-quality structural models. RoseTTAFold has also shown impressive performance in CASP and has been successfully applied to predict the structures of proteins from a wide range of organisms.
A recent study by Krishna et al. [10] expanded on the capabilities of RoseTTAFold by developing RoseTTAFold All-Atom, which includes innovative graphic representations of other biologically relevant molecules in addition to proteins. This advancement leverages a sophisticated neural network architecture to provide precise predictions of protein complex structures, particularly with nucleic acids, small molecule ligands, and post-translational modifications including glycosylations. Similarly, the AlphaFold 3 model, described by Abramson et al. [11], represents another substantial advancement in biomolecular interaction prediction. This model features a diffusion-based architecture capable of joint structure prediction of complexes, including proteins, nucleic acids, small molecules, ions, and modified residues. AlphaFold 3 demonstrates significantly improved accuracy over previous tools, particularly in predicting protein-ligand interactions, protein-nucleic acid interactions, and antibody-antigen predictions. By integrating these capabilities into a unified deep-learning framework, AlphaFold 3 sets a new benchmark for high-accuracy modeling across a diverse range of biomolecular interactions, thus expanding the potential applications in protein modeling and drug design. Table 1 briefly summarizes the key advancement of AI-based structure prediction tools.
Table 1. Comparison of AI-based Protein Structure Prediction Systems
AlphaFold 1 [8] | AlphaFold 2 [5] | RoseTTAFold [6] | ReseTTAFold All-Atom [10] | AlphaFold 3 [11] | |
Developer | Deep Mind | Deep Mind | University of Washington | University of Washington | Deep Mind and Isomorphic Labs |
Key Algorithm | Neural network with distance predictions, gradient descent optimization | Redesigned neural network with Evoformer module and a structure model | 3-track neural network incorporating 1D sequence, 2D distance, and 3D coordinate information | Evolved from RoseTTAFold, graphic representations of small molecule ligands, introduced the all-atom diffusion model | Evolved from AlphaFold 2 with Pairformer module and diffusion model |
Protein Complex Prediction Capability | No | Protein-protein complex (AlphaFold-Multimer) | Protein-protein complex | General biomolecule complex (nucleic acids, ligands, ions, post-translational modifications) | General biomolecule complex (nucleic acids, ligands, ions, post-translational modifications) |
The impact of AI-based protein structure prediction has been further amplified by the development of large-scale structure databases, such as the AlphaFold Protein Structure Database (AlphaFold DB) [12]. The AlphaFold DB, created by DeepMind and EMBL-EBI, contains predicted structures for millions of proteins across various organisms, including a significant portion of the human proteome. As of September 2021, the Protein Data Bank (PDB) contained only about 180,000 experimentally determined structures, covering just over 55,000 distinct proteins [3]. In contrast, the initial release of AlphaFold DB provides over 360,000 predicted structures across 21 proteomes, significantly expanding the structural coverage of the known protein-sequence space [12]. The AlphaFold DB currently contains over 214 million structures [13, 14], which is a massive increase compared to the PDB (Figure 1). The database provides researchers with easy access to high-quality structural models, including those that were previously uncharacterized. The AlphaFold DB has become a valuable resource for the scientific community, enabling researchers to explore the structural basis of protein evolution and function, study protein-protein interactions, and identify potential drug candidates [15].
Figure 1. Comparison of the number of protein structure models in accessible protein database before (2021) and after (current as of June 2024) AI-enabled structure prediction tools were introduced and open to the public.
The combination of state-of-the-art AI-based protein structure prediction methods and comprehensive structure databases has revolutionized the field of structural biology. These advances have opened new avenues for exploring the "dark matter" of the protein universe and have the potential to accelerate the discovery of novel protein families, folds, and functions [16].
3. AI-enabled protein function prediction
Unveiling the functional gems hidden within the dark matter of the protein universe goes beyond structure prediction. AI can be trained to predict protein function based on sequence or structure data. By analyzing known protein sequences and their associated functions, AI models can learn to identify patterns that correlate with specific activities [17]. This opens the door to predicting the function of entirely novel protein sequences, potentially leading to the discovery of proteins with previously unknown applications in medicine, biotechnology, and beyond.
Several studies have demonstrated the power of AI in protein function prediction. For example, DeepFRI [16], a graph convolutional network, predicts protein functions by leveraging sequence features extracted from a protein language model and protein structures. It outperforms current leading methods and scales to the size of current sequence repositories. Another study by Bileschi et al. introduces a deep learning model that learns the language of protein sequences and uses this knowledge to predict protein function directly from the sequence [18]. This AI-enabled study has extended the coverage of Pfam by >9.5%, exceeded additions made over the last decade, and predicted function for 360 human reference proteome proteins with no previous Pfam annotation. This approach demonstrates the potential of AI in this domain.
By rapidly and accurately predicting the functions of uncharacterized proteins, researchers can prioritize targets for experimental validation and further investigation, accelerating the discovery of novel proteins with valuable applications [19].
4. Large-scale exploration of the protein universe
Recent advances in AI-enabled protein structure prediction have led to the generation of structural models for a significant portion of the known protein universe. The AlphaFold Protein Structure Database (AlphaFold DB) contains over 214 million predicted structures, covering most proteins in UniProtKB [12]. This wealth of structural data presents an unprecedented opportunity to explore the "dark matter" of the protein universe, i.e., the vast number of proteins that lack structural and functional annotations.
Two recent studies by Durairaj et al. and Barrio-Hernandez et al. have utilized the AlphaFold DB to investigate the extent of this "dark matter" and uncover novel protein families and folds [13, 14]. Both studies employed large-scale clustering and analysis methods to efficiently process the vast amount of structural data.
Barrio-Hernandez et al. developed a highly scalable structure-based clustering algorithm, Foldseek cluster, capable of handling hundreds of millions of structures [13]. They clustered the entire AlphaFold DB, identifying 2.30 million non-singleton structural clusters. Remarkably, 31% of these clusters lacked annotations, representing probable previously undescribed structures. Although these unannotated clusters covered only 4% of the proteins in the AlphaFold DB, they provide a new resource for studying novel protein families and folds.
Similarly, Durairaj et al. constructed a sequence similarity network of over 6 million UniRef50 clusters with high-confidence AlphaFold models (pLDDT > 90) [14]. They found that 34% of these clusters were functionally "dark," lacking annotations that could provide insights into their biological roles. By exploring this network, they discovered 290 putative new protein families and at least one new protein fold, the β-flower fold. Notably, they experimentally validated a new superfamily of translation-targeting toxin-antitoxin systems, dubbed TumE-TumA.
Both studies showcase the power of combining sequence and structural information to uncover uncharted areas in the protein universe. Durairaj et al. identified the β-flower fold, a symmetric β-barrel structure reminiscent of the Tubby C-terminal domain, and added several new Pfam families based on their analyses [14]. Barrio-Hernandez et al. used structural comparisons to predict domain families and their relationships, identifying examples of remote structural similarity that expand the evolutionary coverage of previously known families [13]. For instance, they found human immune-related proteins with putative remote homology in prokaryotic species, illustrating the potential for cross-kingdom evolution of immunity-related proteins.
The experimental validation of the TumE-TumA toxin-antitoxin system by Durairaj et al. underscores the importance of combining computational predictions with wet lab experiments to verify newly discovered protein families [14]. Such collaborative efforts between computational and experimental biologists will be crucial in unraveling the biological roles of the "dark matter" proteins.
These studies have significant implications for understanding protein function and evolution. By shedding light on the uncharted regions of the protein universe, they pave the way for discovering novel enzymatic activities, regulatory mechanisms, and structural scaffolds. The identification of remote homologies and evolutionary connections across kingdoms can provide insights into the emergence and diversification of protein families. Moreover, the characterization of "dark matter" proteins may lead to new biotechnological and biomedical applications, such as the development of novel biocatalysts, antimicrobial agents, or therapeutic targets.
5. AI-enabled de novo protein design
The power of AI extends beyond discovery and into the realm of protein design. De novo protein design, the creation of proteins with desired functions, has traditionally been a laborious and hit-or-miss process [20]. However, AI-driven design utilizes powerful algorithms to iteratively refine protein sequences, rapidly converging on structures with the targeted properties [21]. This has the potential to revolutionize fields like medicine, where designer proteins could be used to create novel drugs or targeted therapies. Additionally, AI-designed proteins could find applications in materials science, bioremediation, and other areas.
Recent studies have showcased the capabilities of AI in de novo protein design. For example, Anishchenko et al. [22] developed a method using deep network hallucination to create novel proteins with sequences unrelated to naturally occurring ones. They utilized the trRosetta structure prediction network to generate starting residue-residue distance maps from random amino acid sequences. Through Monte Carlo sampling and optimization, they produced diverse protein sequences and structures. The synthetic genes encoding these sequences were expressed in E. coli, and several proteins folded into stable structures consistent with the hallucinated models, demonstrating the potential of deep networks to design new, functional proteins (Figure 2A).
Another study by Watson et al. [23] introduced a novel approach called RFdiffusion, leveraging fine-tuned RoseTTAFold for protein structure denoising tasks. This method enables the generation of diverse protein structures and functional designs, including monomer designs, symmetric oligomers, and complex enzyme active site scaffolds. RFdiffusion's capabilities were demonstrated through experimental validations such as Cryo-EM structure elucidation, confirming the designed structures' accuracy and functionality (Figure 2B).
Recently, exciting progress was made in AI-enabled de novo design of small molecule binders, which used the RFdiffusion All-atom model to generate protein structures that can bind diverse small molecules [10]. The design process started with random distributions of residues around the target small molecules and employed iterative denoising to create coherent protein backbones with complementary pockets. Following sequence design using LigandMPNN, Rosetta GALigandDock energy calculations were used to evaluate the protein-small molecule interface and AlphaFold 2 predictions to evaluate the extent to which the sequence encodes the designed structure. Experimental characterization of these binders demonstrated successful binding to small molecule ligands such as digoxigenin and heme. For digoxigenin, the highest affinity binder showed a Kd of 343 nM and high thermostability. For heme binders, 90 out of 168 designs had UV/Vis spectra consistent with Cys-bound heme, with 33 experimentally purified as monomeric proteins and showing heme-binding in size exclusion chromatography. For one representative protein, HEM_3.C9, the crystal structure was determined and the accuracy of the designed protein structure was confirmed (Figure 2C). This integrated approach shows a significant advancement in the rational de novo design of protein binders to small molecules.
The progress demonstrates the potential of AI in designing proteins with desired properties, pushing the boundaries of what is possible in protein engineering. As AI-driven design methods continue to advance, we can expect to see an increasing number of de novo designed novel proteins with tailored functions, further expanding the boundary of protein universe in biotechnology and medicine.
|
|
|
A | B | C |
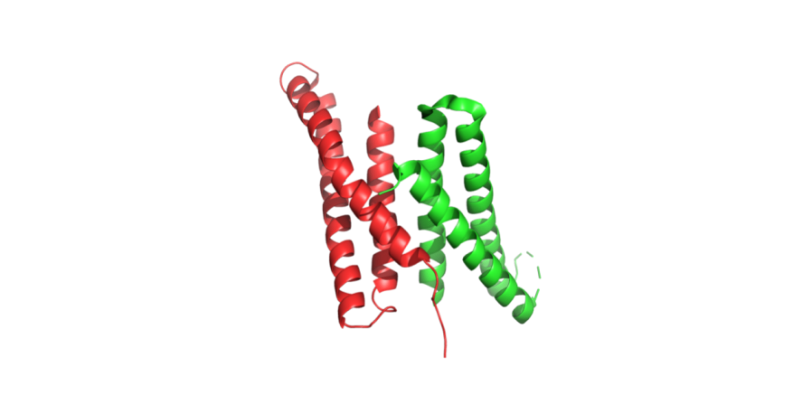

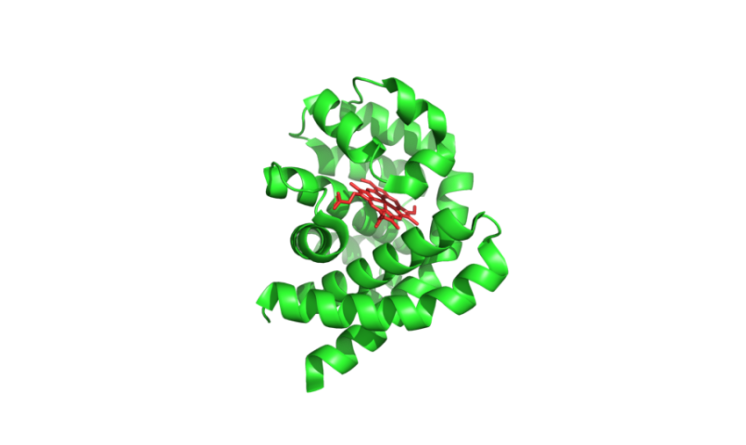
Figure 2. Experimental determination of structures of de novo designed proteins, which demonstrate closely matches of the AI-designed models. (A) Crystal structure (PDB# 7K3H) of a de novo designed protein dimer, 0217, with each subunit shown in green and red, respectively. (B) Cryo-EM structure (PDB# 8SK7) of a de novo designed Influenza HA binder, HA_20 (shown in red), bound to Influenza HA (shown in green). (C) Crystal structure (PDB# 8VC8) of a de novo designed heme-binding protein HEM_3.C9 (shown in green), in complex with heme (shown in red).
6. Challenges and future directions
While AI-enabled exploration of the protein universe has made remarkable progress, significant challenges remain in refining these methods, integrating predicted structures with experimental data, and leveraging novel protein families and folds for various applications. Moreover, a substantial portion of the protein universe remains as "dark matter," requiring innovative approaches for annotation and characterization.
One of the primary challenges is to continue refining AI-enabled structure prediction methods to improve their accuracy and robustness. Incorporating additional constraints from physics-based modeling, co-evolutionary analysis, and experimental data could help overcome limitations, particularly for proteins with unique structural features, intrinsically disordered regions, transmembrane regions, or limited evolutionary information [24, 25]. Further optimizing and developing methods that can accurately and reliably predict protein-protein interactions, protein-ligand binding, and the impact of mutations on structure and function will greatly expand the utility of these tools [26].
Another important challenge is the experimental validation of AI-generated predictions and designs. While AI methods can rapidly generate hypotheses about protein structure, function, and design, these predictions must be verified through experimental studies. Developing high-throughput methods for protein expression, purification, and characterization will be essential to keep pace with the growing number of AI-generated predictions and designs. Collaborative efforts between computational and experimental researchers will be crucial in this regard, ensuring that the most promising leads are prioritized for validation and further investigation [24].
7. Conclusion
AI-enabled exploration of the protein universe has made remarkable progress. By integrating AI-driven protein structure prediction, function prediction, annotation, and de novo design, researchers have illuminated the dark matter of the protein universe and unlocked its potential for basic biology and applied sciences. However, significant challenges and opportunities lie ahead for future research. Enhancements in AI algorithms are needed to improve accuracy in predicting structures and dynamic movements of proteins and protein complexes. Additionally, there's a pressing need for high-throughput, cost-effective experimental techniques to validate the vast number of AI-generated hypotheses. Potential solutions include the use of advanced automation and robotic technologies in laboratories. Looking forward, as we continue to push the boundaries of our understanding of protein universe, we can anticipate more transformative advances driven by the synergy between AI and protein science, which can revolutionize our understanding of basic biology and enhance our ability to engineer novel biological materials and systems.
References
[1]. Akdel, M., et al., A structural biology community assessment of AlphaFold2 applications. Nat Struct Mol Biol, 2022. 29(11): p. 1056-1067.
[2]. Jaroszewski, L., et al., Exploration of uncharted regions of the protein universe. PLoS Biol, 2009. 7(9): p. e1000205.
[3]. Perrakis, A. and T.K. Sixma, AI revolutions in biology: The joys and perils of AlphaFold. EMBO Rep, 2021. 22(11): p. e54046.
[4]. Lam, S.D., et al., An overview of comparative modelling and resources dedicated to large-scale modelling of genome sequences. Acta Crystallogr D Struct Biol, 2017. 73(Pt 8): p. 628-640.
[5]. Jumper, J., et al., Highly accurate protein structure prediction with AlphaFold. Nature, 2021. 596(7873): p. 583-589.
[6]. Baek, M., et al., Accurate prediction of protein structures and interactions using a three-track neural network. Science, 2021. 373(6557): p. 871-876.
[7]. Orengo, C.A. and J.M. Thornton, Protein families and their evolution-a structural perspective. Annu Rev Biochem, 2005. 74: p. 867-900.
[8]. Senior, A.W., et al., Improved protein structure prediction using potentials from deep learning. Nature, 2020. 577(7792): p. 706-710.
[9]. Pereira, J., et al., High-accuracy protein structure prediction in CASP14. Proteins, 2021. 89(12): p. 1687-1699.
[10]. Krishna, R., et al., Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science, 2024. 384(6693): p. eadl2528.
[11]. Abramson, J., et al., Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 2024.
[12]. Varadi, M., et al., AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res, 2022. 50(D1): p. D439-D444.
[13]. Barrio-Hernandez, I., et al., Clustering predicted structures at the scale of the known protein universe. Nature, 2023. 622(7983): p. 637-645.
[14]. Durairaj, J., et al., Uncovering new families and folds in the natural protein universe. Nature, 2023. 622(7983): p. 646-653.
[15]. Bryant, P., G. Pozzati, and A. Elofsson, Improved prediction of protein-protein interactions using AlphaFold2. Nat Commun, 2022. 13(1): p. 1265.
[16]. Gligorijevic, V., et al., Structure-based protein function prediction using graph convolutional networks. Nat Commun, 2021. 12(1): p. 3168.
[17]. Zhang, F., et al., DeepFunc: A Deep Learning Framework for Accurate Prediction of Protein Functions from Protein Sequences and Interactions. Proteomics, 2019. 19(12): p. e1900019.
[18]. Bileschi, M.L., et al., Using deep learning to annotate the protein universe. Nat Biotechnol, 2022. 40(6): p. 932-937.
[19]. Bugnon, L.A., et al., Transfer learning: The key to functionally annotate the protein universe. Patterns (N Y), 2023. 4(2): p. 100691.
[20]. Huang, P.S., S.E. Boyken, and D. Baker, The coming of age of de novo protein design. Nature, 2016. 537(7620): p. 320-7.
[21]. Kortemme, T., De novo protein design-From new structures to programmable functions. Cell, 2024. 187(3): p. 526-544.
[22]. Anishchenko, I., et al., De novo protein design by deep network hallucination. Nature, 2021. 600(7889): p. 547-552.
[23]. Watson, J.L., et al., De novo design of protein structure and function with RFdiffusion. Nature, 2023. 620(7976): p. 1089-1100.
[24]. Versini, R., et al., A Perspective on the Prospective Use of AI in Protein Structure Prediction. J Chem Inf Model, 2024. 64(1): p. 26-41.
[25]. Tamburrini, K.C., et al., Predicting Protein Conformational Disorder and Disordered Binding Sites. Methods Mol Biol, 2022. 2449: p. 95-147.
[26]. Tsaban, T., et al., Harnessing protein folding neural networks for peptide-protein docking. Nat Commun, 2022. 13(1): p. 176.
Cite this article
Song,E.Z. (2024). AI-enabled exploration of the "dark matter" of the protein universe. Theoretical and Natural Science,59,85-92.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 4th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Akdel, M., et al., A structural biology community assessment of AlphaFold2 applications. Nat Struct Mol Biol, 2022. 29(11): p. 1056-1067.
[2]. Jaroszewski, L., et al., Exploration of uncharted regions of the protein universe. PLoS Biol, 2009. 7(9): p. e1000205.
[3]. Perrakis, A. and T.K. Sixma, AI revolutions in biology: The joys and perils of AlphaFold. EMBO Rep, 2021. 22(11): p. e54046.
[4]. Lam, S.D., et al., An overview of comparative modelling and resources dedicated to large-scale modelling of genome sequences. Acta Crystallogr D Struct Biol, 2017. 73(Pt 8): p. 628-640.
[5]. Jumper, J., et al., Highly accurate protein structure prediction with AlphaFold. Nature, 2021. 596(7873): p. 583-589.
[6]. Baek, M., et al., Accurate prediction of protein structures and interactions using a three-track neural network. Science, 2021. 373(6557): p. 871-876.
[7]. Orengo, C.A. and J.M. Thornton, Protein families and their evolution-a structural perspective. Annu Rev Biochem, 2005. 74: p. 867-900.
[8]. Senior, A.W., et al., Improved protein structure prediction using potentials from deep learning. Nature, 2020. 577(7792): p. 706-710.
[9]. Pereira, J., et al., High-accuracy protein structure prediction in CASP14. Proteins, 2021. 89(12): p. 1687-1699.
[10]. Krishna, R., et al., Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science, 2024. 384(6693): p. eadl2528.
[11]. Abramson, J., et al., Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 2024.
[12]. Varadi, M., et al., AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res, 2022. 50(D1): p. D439-D444.
[13]. Barrio-Hernandez, I., et al., Clustering predicted structures at the scale of the known protein universe. Nature, 2023. 622(7983): p. 637-645.
[14]. Durairaj, J., et al., Uncovering new families and folds in the natural protein universe. Nature, 2023. 622(7983): p. 646-653.
[15]. Bryant, P., G. Pozzati, and A. Elofsson, Improved prediction of protein-protein interactions using AlphaFold2. Nat Commun, 2022. 13(1): p. 1265.
[16]. Gligorijevic, V., et al., Structure-based protein function prediction using graph convolutional networks. Nat Commun, 2021. 12(1): p. 3168.
[17]. Zhang, F., et al., DeepFunc: A Deep Learning Framework for Accurate Prediction of Protein Functions from Protein Sequences and Interactions. Proteomics, 2019. 19(12): p. e1900019.
[18]. Bileschi, M.L., et al., Using deep learning to annotate the protein universe. Nat Biotechnol, 2022. 40(6): p. 932-937.
[19]. Bugnon, L.A., et al., Transfer learning: The key to functionally annotate the protein universe. Patterns (N Y), 2023. 4(2): p. 100691.
[20]. Huang, P.S., S.E. Boyken, and D. Baker, The coming of age of de novo protein design. Nature, 2016. 537(7620): p. 320-7.
[21]. Kortemme, T., De novo protein design-From new structures to programmable functions. Cell, 2024. 187(3): p. 526-544.
[22]. Anishchenko, I., et al., De novo protein design by deep network hallucination. Nature, 2021. 600(7889): p. 547-552.
[23]. Watson, J.L., et al., De novo design of protein structure and function with RFdiffusion. Nature, 2023. 620(7976): p. 1089-1100.
[24]. Versini, R., et al., A Perspective on the Prospective Use of AI in Protein Structure Prediction. J Chem Inf Model, 2024. 64(1): p. 26-41.
[25]. Tamburrini, K.C., et al., Predicting Protein Conformational Disorder and Disordered Binding Sites. Methods Mol Biol, 2022. 2449: p. 95-147.
[26]. Tsaban, T., et al., Harnessing protein folding neural networks for peptide-protein docking. Nat Commun, 2022. 13(1): p. 176.