







1. Introduction
Cancer accounts for approximately 10 million fatalities annually, making it the primary non-natural cause of death globally [1]. In light of the considerable side effects and the resistance often encountered with conventional treatments, cellular immunotherapy is emerging as an increasingly favored alternative for various cancer types.
CAR technology was initially implemented in T lymphocytes, and to date, CAR-T therapies have exhibited remarkable therapeutic potency in the management of specific hematologic malignancies, including acute lymphoblastic leukemia, mantle cell lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, and multiple myeloma [2]. However, the limited infiltration of CAR-T into solid tumor masses, the deactivation of CAR-T within the immunosuppressive TME, and the tendency to cause side effects like CRS mean that the effectiveness of CAR-T therapies in solid tumors is still below ideal [3]. The importance of the innate immune response as the first line of defense for tumor surveillance and elimination is underscored by these challenges, which also highlight the basic shortcomings of strategies that solely concentrate on the adaptive immune system.
In contrast to the extensive scholarly discourse surrounding CAR-T, the evolution of CAR-NK and CAR-M-based treatments is still in its nascent phase, with a paucity of systematic and thorough scholarly inquiry. By analyzing the fundamental mechanisms of action and the most recent clinical uses of CAR-T, the current manuscript aims to critically evaluate the limitations and difficulties existing in CAR-T modalities. In an effort to overcome the obstacles encountered with CAR-T interventions in the context of solid tumor therapy, this review will delineate the comparative advantages of CAR-NK and CAR-M over CAR-T and assess their viability as potential alternative therapeutic strategies. Ultimately, we will investigate collaborative interactions among these three distinct populations of CARs as a promising optimization strategy to augment the therapeutic potency of CAR-mediated cellular immunotherapies.
2. CAR-T
2.1. Structure and Mechanism of CAR-T
2.1.1. Structure of CAR-T
As shown in figure 1, according to some research, the expression and signaling threshold of CARs in CAR-T can also be altered by the structure of the hinge and transmembrane sections [4]. The cytoplasmic signaling domain activates CAR-T by sending downstream signals, which eventually cause tumor cells to become cytotoxic.
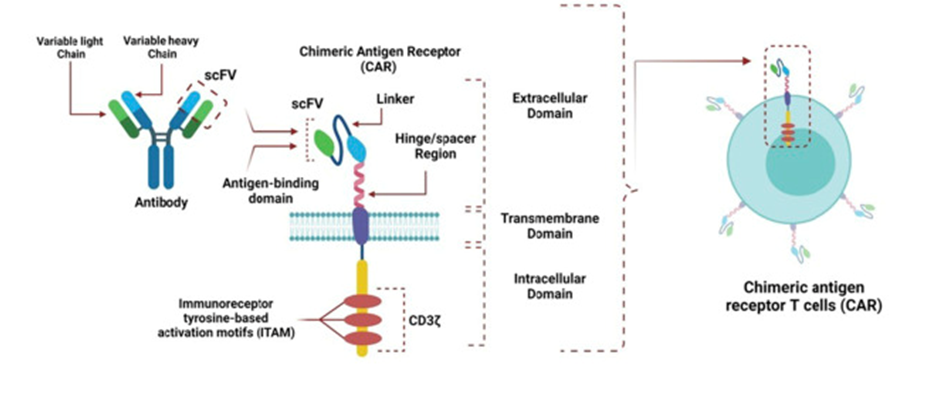
Figure 1: the structure of CAR-T [5].
2.1.2. Mechanism of CAR-T
Anti-tumor mechanisms of this method, which include the death receptor pathway, the perforin-granzyme pathway, and cytokine production, are comparable to those of natural effector T cells. After identifying the tumor antigens, CAR-T use either the death receptor route or the perforin-granzyme pathway to cause cytotoxicity in tumor cells. Antigen-Presenting Cells (APCs) phagocytose the newly released tumor antigens (epitope spreading) by apoptotic tumor cells. Endogenous T cells are exposed to the novel antigens by these APCs. Through their interactions with other immune cells and the tumor stroma, CAR-T cytokine release facilitates tumor destruction [5].
2.2. Clinical Trials of CAR-T
2.2.1. Neuroblastoma(NB)
GD2 is a cell surface marker of NB. In a study designed to test the feasibility and safety of a sort of autologous 1RG-CAR-T (anti-GD2 CAR-T) therapy in patients with relapsed or refractory NB, two of six patients treated with GD2 CAR-T experienced grade 2-3 CRS. Soft tissue degeneration occurred in 3 cases. The conclusion of this trial indicates that more modification of GD2 CAR-T is needed to achieve better efficacy.
2.2.2. Prostate Cancer(PCa)
Around the world, PCa accounts for 375,304 fatalities and 1,414,259 new cases, making it the second most frequent cancer in males. Five patients received treatment with first-generation PSMA CAR-T in a clinical trial that has been completed (NCT00664196). While there was evidence of disease progression, two of the five patients experienced PR. The ineffective CAR-T design in terms of T cell multiplication and recruitment may be the cause of this result [6].
Seven patients received second-generation anti-PSMA CAR-T in a different phase I/II dose-escalation study (NCT01140373). One × 109 cells/kg were given to four patients. Additionally, injections of 1.5 to 3 × 109 cells/kg were given to the remaining patients. Two groups of patients had radiographic SD for longer than six and sixteen months, respectively. The results of the study indicated that all patients experienced disease deterioration. In addition, patients in the second group all presented mild self-limited CRS [7].
2.3. Limitations and Challenges of CAR-T
The findings from the aforementioned clinical trials indicate that while CAR-T has made significant strides in addressing hematological cancers, it continues to encounter numerous limitations and obstacles when applied to solid tumors.
2.3.1. Inhibition of Tumor Microenvironment(TME)
The solid TME is rife with numerous immunosuppressive factors, including regulatory T cells (Tregs), Myeloid-Derived Suppressor Cells (MDSCs), and Tumor-Associated Macrophages (TAMs), all of which can dampen the effectiveness of CAR-T. Additionally, the low-oxygen and acidic conditions prevalent in the TME further compromise the viability and performance of CAR-T.
2.3.2. Permeability and distribution of CAR-T
The complex structure and poor angiogenesis of solid tumors make it difficult for CAR-T to effectively penetrate into the tumor. In addition, fibrosis and stromal barriers within the tumor can also hinder CAR-T migration.
2.3.3. Antigen heterogeneity and escape of CAR-T
Antigenic heterogeneity, or the expression of distinct antigens by distinct tumor cells, is typically prominent in solid tumors. This makes using a single antigen-targeted CAR-T to eradicate all tumor cells challenging. Treatment failure can also result from tumor cells' ability to resist immune attack by down-regulating or deleting antigens that CAR-T target.
2.3.4. Persistence and functional depletion of CAR-T
The short-lived therapy impact may result from the poor endurance of CAR-T in solid tumors when compared to hematologic malignancies. The anti-tumor effectiveness of CAR-T may be impacted by functional exhaustion brought on by prolonged activation in the TME, which is reflected in the increased expression of inhibitory receptors including PD-1 and TIM-3.
3. CAR-NK
CAR-Natural Killer (CAR-NK) cells are genetically engineered NK cells to enhance their ability to recognize and kill specific tumor cells.
3.1. SCAR-NK
CAR-NK and CAR-T are similar in that they introduce receptors that recognize tumor-specific antigens (TSAs) into NK cells, enabling them to recognize and kill tumor cells with greater efficiency.
3.1.1. Structure of CAR-NK
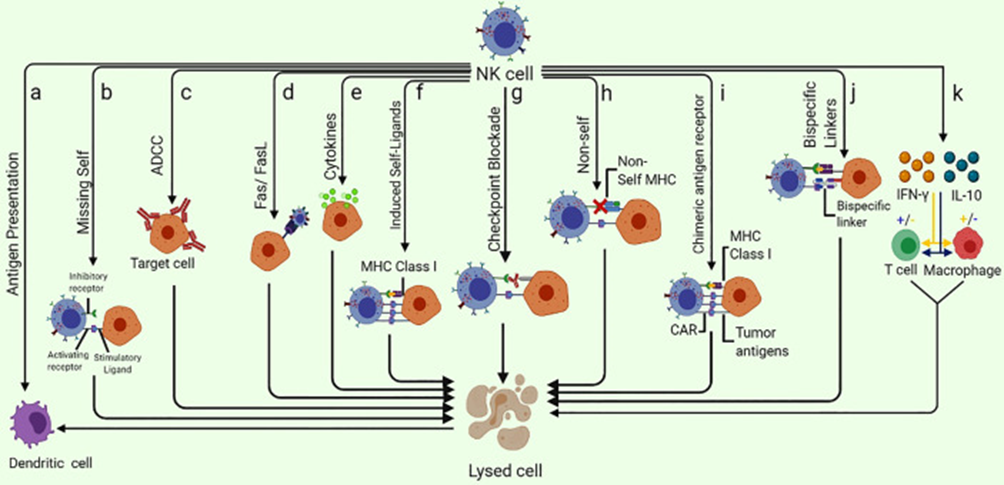
Figure 2: Different ways of NK-cell mediated tumor killing and immune system modulation [5].
Different ways of NK-cell mediated tumor killing and immune system modulation [5].
A shown in figure 2, CARs' antitumor activity can be increased by redesigning them with NK cell signaling-associated domains. When compared to CAR-T constructions, CARs that include NK cell-specific co-stimulatory domains such as 2B4, DAP-10, or DAP-12 have demonstrated increased cytotoxicity and IFN-γ secretion.
3.1.2. Mechanism
Compared to CAR-T, CAR-NK exhibits unique advantages in the treatment of solid tumors, including reduced toxicity and improved safety. The following mechanisms describe how CAR-NKs act on solid tumors.
CAR-NKs specifically recognize and bind to particular antigens on the surface of tumor cells via their surface CAR domains. This binding is not restricted by the MHC, allowing CAR-NK to be more broadly applicable to various types of tumors. Activated CAR-NK can kill tumor cells through multiple mechanisms, primarily by releasing perforin and granzyme B to directly disrupt the tumor cell membrane (Figure 2).
Some studies suggest that CAR-NK may possess a certain degree of memory characteristics, enabling a faster and stronger response upon reencountering the same antigen after the initial exposure, thus providing long-term protective effects. Additionally, CAR-NK can overcome immunosuppressive factors present in the TME by secreting anti-inflammatory cytokines or expressing related receptors, thereby enhancing therapeutic efficacy.
3.2. Progressive Advantages of CAR-NK
CAR-NK are genetically engineered NKs to enhance their ability to recognize and kill specific tumor cells.
3.2.1. Lower toxicity and higher safety
A significant advantage of CAR-NK is its lower toxicity and improved safety profile. Compared with CAR-T, the CRS and neurotoxicity caused by CAR-NK are milder. This is because NKs themselves have low levels of cytokine secretion and a relatively short duration in the body, reducing the risk of excessive activation and persistent inflammatory responses. Furthermore, the CAR-NK were derived from allogeneic donors and thus did not cause severe Graft-versus-Host Disease (GvHD). This feature makes this method safer in clinical applications, especially when multiple infusions are required [8].
3.2.2. More antigen receptors
CAR-NK can target a variety of tumor-associated surface antigens, including some antigens that are difficult to target by CAR-T. For example, CAR-NK can effectively target HER2, NKG2D ligands, CEA and other antigens expressed in a variety of solid tumors. This makes CAR-NK more promising for the treatment of multiple types of solid tumors.
3.2.3. Enhanced TME adaptability
The function of CAR-T is negatively impacted by the immunosuppressive characteristics of the TME of solid tumors. CAR-NK, on the other hand, is more adaptable to the TME. Through a variety of strategies, such as luring other immune cells with cytokines and chemokines and eliminating tumor cells through direct contact, NK cells are able to counteract the TME's suppressive effects.
3.2.4. Shorter preparation time and higher accessibility
Because CAR-NK often produces more quickly than CAR-T, CAR-NK treatment is more adaptable and available for clinical use. Furthermore, NK cells can be derived from a number of sources, such as peripheral blood, umbilical cord blood, induced pluripotent stem cells (iPSCs), etc., which makes it possible to produce them on a large scale and prepare them consistently [9].
3.3. Clinical Trials of CAR-NK
As an emerging immunotherapy method, CAR-NK has shown significant potential in the treatment of a variety of malignant solid tumors. Compared with conventional CAR-T, CAR-NK has a lower toxicity profile and a higher safety profile, making it an attractive treatment option.
3.3.1. Glioblastoma Multiforme (GBM)
GBMs have the most extensive preclinical data available for CAR-N. Related studies have shown that about 89% of GBM are able to be naturally infiltrated by NKs, and in a study of intravenous or intratumorally injection of unmodified activated autologous NKs into 9 GBM patients, 4 patients achieved partial/mixed response to the tested treatment and 3 patients achieved stable disease [10].
3.3.2. Pancreatic Cancer (PC)
Immunosuppressive stroma is prevalent in pancreatic ductal adenocarcinoma (PDAC), making up as much as 70% of the tumor volume [11]. Folate Receptor α (FRα) and Death Receptor 4/5 (DR4/5) were shown to be the most promising targets for CAR-NK treatment in a study that increased PDAC-specific membrane markers.
Furthermore, MUC1-directed CAR-NKs were examined in a variety of cancers in the preclinical context. CAR constructs that target MUC1 and PD-1 in CAR-NK were discovered in eight different cancer types that tested positive for both PD-1 and MUC1, including ovarian, lung, pancreatic, and colon cancers, with seven patients exhibiting stable disease. Furthermore, the study found that dual-targeted treatment did not cause any significant side effects [12].
3.3.3. Breast Cancer (BC)
CAR-NKs have also shown potential in the treatment of BC. One study used HER2-specific CAR-NK to treat HER2-positive BC patients, and the results showed that CAR-NK could effectively kill tumor cells with low toxic side effects.
4. CAR-M
CAR-M is an emerging cell therapy, which introduces CARs that recognize specific tumor-associated antigens (TAAs) into macrophages through genetic engineering technology, so that these macrophages can specifically target and phagocytose tumor cells. As a part of the immune system, macrophages have strong phagocytic capacity and the ability to regulate the surrounding microenvironment, so they have shown great potential in cancer therapy.
4.1. Structure and Mechanism of CAR-M
4.1.1. Structure of CAR-M
CAR-M and first-generation CAR-T have only minor differences in their antigen-binding domain, hinge region, and transmembrane domain [10]. However, CAR-M exhibit greater diversity in their cytoplasmic signaling domains. The cytoplasmic domain of CD3ζ shares significant homology with the natural Fc Receptor γ chain (FcRγ) [13]. Consequently, the activation domains of CD3ζ and FcRγ are the most commonly used cytoplasmic signaling domains in CAR-M, capable of triggering effective antigen-specific phagocytosis.
In addition to these, researchers have also explored other unique signaling domains in CAR-M, such as the activation domain of CD147. The expression and secretion of Matrix Metalloproteinases (MMPs) is upregulated by this signaling domain, which does not trigger phagocytosis. The dense Extracellular Matrix (ECM) surrounding solid tumors can be degraded by MMPs, which allows immune cells to infiltrate the tumor site [14]. Tyrosine protein kinase Mer (MerTK), multiple epidermal growth factor-like domains protein 10 (Megf10), and toll-like receptors [15] can also be included as optional signaling domains that stimulate phagocytosis or the release of pro-inflammatory cytokines. The anti-tumor mechanisms that CAR-Ms execute are made possible by their diverse cytoplasmic signaling domains.
4.1.2. Mechanism of CAR-M
The antitumor mechanisms of macrophages are similar to those of endogenous macrophages. However, a distinct advantage of CAR-M over CAR-T arises from its unique indirect antitumor mechanisms in solid tumors. The interaction between CAR-Ms and other immune cells is tightly regulated. For instance, in vivo, CAR-M recruit T cells to the tumor site by secreting chemokines. CAR-M can still present tumor antigens to T cells while maintaining their APC capabilities when T cells arrive. Moreover, the expression levels of CD80/CD86 on CAR-M are significantly higher than those on natural macrophages, thereby not only enhancing the activity of the antitumor T cell response but also improving its infiltration and persistence [16].
4.2. Progressive Advantages of CAR-M
CAR-M has shown many advantages over CAR-T in the treatment of solid tumors.
4.2.1. Lower toxicity and higher safety
CAR-M, like CAR-NK, has lower toxicity and higher safety than CAR-T. The CRS and neurotoxicity caused by CAR-M are mild, mainly because macrophages have lower levels of cytokine secretion in vivo and have a relatively short duration in vivo, reducing the risk of excessive activation and persistent inflammation.
Moreover, CAR-M can be obtained from healthy donors, which opens up the possibility of allogeneic use. Unlike CAR-T, CAR-M cause a lower risk of GvHD in allogeneic application, which makes CAR-M safer and more feasible in clinical applications.
4.2.2. Powerful phagocytosis and antigen presentation
CAR-M have strong phagocytic ability and antigen presentation function, which makes them more effective in recognizing and eliminating tumor cells in the TME. In addition, CAR-M can phagocytose tumor cells to present tumor antigens, activate T cells, and form a synergistic immune effect to enhance the overall anti-tumor immune response.
4.2.3. Versatility
CAR-Ms have versatility. They can not only directly kill tumor cells, but also regulate the immune microenvironment by secreting cytokines and chemokines to promote the activation and recruitment of other immune cells. This multiple mechanism of action makes CAR-M uniquely advantageous in the treatment of complex and heterogeneous solid tumors.
4.3. Clinical Trials of CAR-M
4.3.1. BC
CAR-M have demonstrated considerable promise in combating BC. A phase I study evaluated the safety and initial effectiveness of a HER2-targeted CAR-M (CT-0508) in individuals diagnosed with HER2-positive BC. Findings indicated that CAR-M were able to effectively gather within the TME produce anti-tumor responses, while maintaining manageable side effects [17].
4.3.2. Gastric cancer (GC)
CAR-M for GC is also being actively explored. A study using CAR-M targeting Claudin 18.2 showed a good safety profile and preliminary antitumor activity in patients with GC. In addition, CAR-M can effectively improve the TME and promote the infiltration and activation of other immune cells [18].
4.3.3. PC
PC is a highly malignant tumor, which is difficult to treat. CAR-M has also shown potential in the treatment of PC. A preclinical study showed that EGFR-targeting CAR-M significantly inhibited tumor growth and prolonged survival in a mouse model of PC.
5. CAR Cells Combination Therapy
5.1. Synergistic Effect of CAR-T and CAR-NK
The combination of CAR-T and CAR-NK can lead to an improvement in functionality. This combined therapy can bridge the gaps between CAR therapies and enhance their antitumor efficacy in solid tumors. CAR-T and CAR-NK have complementary roles in antitumor functions. The delayed response of CAR-T and the ability to cope with bursts of adverse reactions can be improved by CAR-NK' rapid cytotoxicity and regulatory processes [19]. In contrast, CAR-T have a longer persistence and better proliferative capacity upon encountering antigens, overcoming the issues of short lifespan and poor proliferation associated with CAR-NK.
A study explored the synergistic effects of combined CAR-T and CAR-NK on MM and NHL models both in vitro and in vivo. In this context, CAR-NK can exhibit immunomodulatory effects on T cells, enhancing their antitumor functionality. High doses of CAR-NK and NKs are effective, as indicated by the results, and adding a small amount of NKs can enhance the early activation of CAR-T in MM cells and improve their migration.
Furthermore, NKs improve the cytokine profile, preventing the occurrence of CRS, while also enhancing the adaptability of CAR-T to prevent exhaustion [20]. This combination regimen has achieved some success in solid tumors. In xenograft TME models, NKG2Dζ chimeric receptor-modified NKs can appropriately enhance the antitumor functionality of CAR-T through the elimination of MDSCs. Additionally, NKG2Dζ-NKs produce pro-inflammatory chemokines and cytokines in response to MDSCs at the tumor site, thereby enhancing the functionality and infiltration of CAR-T.
5.2. Synergistic Effect of CAR-T and CAR-M
CAR-M can generate pro-inflammatory cytokines to improve TME and have the ability to locate and infiltrate solid tumors with possible benefits. Despite the rapid advancements in CAR-T immunotherapy, there are still few clinical responses in solid tumors. Limited penetration into the thick extracellular matrix and immunosuppressive TME depletion are obstacles to CAR-T treatment in solid tumors [21]. Moreover, macrophages can promote the activation of T cells. Therefore, CAR-M and CAR-T together might help improve tumor response.
Research has indicated that inflammatory agents released by CAR-T can trigger M1 polarization in macrophages, boost the expression of co-stimulatory ligands such as CD86 and CD80 on CAR-M, and heighten the cytotoxic capabilities of CAR-M. This increase in co-stimulatory ligands may contribute to the activation and adaptability of CAR-T, showcasing a synergistic interaction between CAR-T and CAR-M in the eradication of tumor cells.
5.3. Synergistic Effect of CAR-M and CAR-NK
CAR-NK and CAR-M have demonstrated distinct benefits in the realm of cancer immunotherapy. Lately, scientists have started investigating the combined potential of CAR-NK and CAR-M to enhance treatment outcomes for solid tumors.
CAR-NK and CAR-M have complementary roles in killing tumor cells. CAR-NK kill tumor cells mainly through cytotoxicity and Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) . In contrast, CAR-M eliminate tumor cells through MHC presentation. The combination of the two can more comprehensively cover the mechanism of tumor cell clearance and improve the therapeutic effect.
CAR-M have the potential to enhance the TME through several mechanisms. By releasing cytokines like IL-12, they can stimulate NKs and various other immune cells, thereby bolstering the body's anti-tumor response. Additionally, CAR-NK contributes to this improvement by secreting a range of cytokines and chemokines, which facilitate the activation and recruitment of other immune cells. The combined action of these two types of cells can more effectively transform the TME and strengthen the overall immune response against tumors.
The combination of CAR-NK and CAR-M can enhance immune memory and improve the long-term anti-tumor effect. CAR-M activates T cells through antigen presentation function and forms immune memory, while CAR-NK maintains tumor surveillance through continuous cytotoxic effects. This synergistic effect helps to prevent tumor recurrence and metastasis.
At present, the research on the combination of CAR-M and CAR-NK in the treatment of solid tumors is still in the early stages, and most studies focus on laboratory and preclinical models. However, there are preliminary data indicating that this combination therapy shows promising efficacy and safety in certain solid tumor models.
In a study utilizing a mouse model, researchers found that CAR-NK were effective in treating non-small-cell lung cancer, as they notably inhibited tumor growth and extended the survival of the subjects. Interestingly, when CAR-NK were paired with CAR-M, the anti-tumor effect was even more pronounced. Other studies have shown that the combined treatment of CAR-M and CAR-NK can effectively inhibit tumor growth in BC xenograft models and reduce the number of metastatic lesions [22].
6. Discussion
This review primarily investigates the feasibility of introducing CAR-NK and CAR-M as alternative therapies to CAR-T to enhance the efficacy of solid tumor treatment. However, beyond this, there are other methods to improve the effectiveness of CAR-T against solid tumors, such as altering the design of CAR-T to produce matrix-degrading enzymes that break down the physical barriers in solid tumors, thereby enhancing the migration and infiltration capabilities of CAR-T; or by administering CAR-T via intraperitoneal or intratumorally injection to increase their accumulation at the tumor site.
Currently, CAR-NK and CAR-M therapies are still in their infancy. Despite their advantages in the treatment of solid tumors, CAR-T and other therapies still have some limitations compared to each other. For instance, CAR-NK has lower transduction efficiency and persistence in peripheral blood and tissues; CAR-Ms lack suitable TSA and TAAs, and they are prone to antigen escape. Future research can promote the broader application of CAR-NK and CAR-M therapies by optimizing the structure and manufacturing of CARs, identifying new targeting antigens, and other means.
7. Conclusion
In order to treat solid tumors, we examined the current research state of CAR-T, CAR-NK, and CAR-M. We found that these three adoptive cell therapies differed significantly in terms of structure, anticancer mechanisms, and clinical trial advancement. After examining these variations, we came to the conclusion that CAR-NK and CAR-M therapies can successfully handle some of the problems that CAR-T presents when treating solid tumors. CAR-NKs are less expensive than CAR-T and can lower the risk of neurotoxicity and CRS; CAR-Ms are more successful at controlling solid tumors because they have a wider variety of antitumor mechanisms and more potent tumor infiltration capabilities. As a result, they can be employed as CAR-T substitutes and are more frequently used to treat solid tumors. Simultaneously, we discovered that CAR-M can be highly effective against tumors when combined with CAR-NK or CAR-T.
Authors Contribution
All the authors contributed equally and their names were listed in alphabetical order.
References
[1]. Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., & Bray, F. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 71(3), 209–249. https://doi.org/10.3322/caac.21660
[2]. Huo, Y., Zhao, X., Wang, X., Zhang, H., Wang, X., Zhou, L., & Zhang, H. (2023). M1 polarization enhances the antitumor activity of chimeric antigen receptor macrophages in solid tumors. Journal of Translational Medicine, 21(1), 225. https://doi.org/10.1186/s12967-023-04206-z
[3]. Shin, M. H., Lee, H. J., & Kwon, J. (2023). Recent advances in CAR-based solid tumor immunotherapy. Cells, 12(12), 1606. https://doi.org/10.3390/cells12121606
[4]. Alnefaie, A., Albogami, S., Asiri, Y., Ahmad, T., Alotaibi, S. S., Al-Sanea, M. M., & Althobaiti, H. (2022). Chimeric Antigen Receptor T-Cells: An overview of concepts, applications, limitations, and proposed solutions. Frontiers in Bioengineering and Biotechnology, 10, 797440. https://doi.org/10.3389/fbioe.2022.797440
[5]. Straathof, K., Hough, R., & Xie, F. (2020). Antitumor activity without on-target off-tumor toxicity of GD2–chimeric antigen receptor T cells in patients with neuroblastoma. Science Translational Medicine, 12(571), eabd6169. https://doi.org/10.1126/scitranslmed.abd6169
[6]. Junghans, R. P., & Nguyen, L. (2016). Phase I trial of anti‐PSMA designer CAR‐T cells in prostate cancer: Possible role for interacting interleukin 2‐T cell pharmacodynamics as a determinant of clinical response. The Prostate, 76(14), 1257–1270. https://doi.org/10.1002/pros.23212
[7]. Han, J., Zhang, X., & Wang, J. (2015). CAR-engineered NKs targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Scientific Reports, 5, 11483. https://doi.org/10.1038/srep11483
[8]. Wrona, E., Borowiec, M., & Potemski, P. (2021). CAR-NKs in the treatment of solid tumors. International Journal of Molecular Sciences, 22(11), 5899. https://doi.org/10.3390/ijms22115899
[9]. Köylijärvi, M., Koskinen, K., & Jäntti, J. (2020). Translation of small-scale CAR-T cell manufacturing methods to a clinical-scale production platform. Cytotherapy, 22(5), S127–S128. https://doi.org/10.1016/j.jcyt.2020.02.107
[10]. Zhang, L., & Liu, C. (2022). Natural Killer Cell-Based Cancer Immunotherapy: From Bench to Bedside. In Natural Killer Cells: Lessons and Challenges (pp. 25–48). Springer. https://doi.org/10.1007/978-3-030-63271-9_2
[11]. Rossi, F., Pappalardo, S., & Fava, L. (2022). Next generation natural killer cells for cancer immunotherapy. Frontiers in Immunology, 13, 886429. https://doi.org/10.3389/fimmu.2022.886429
[12]. Kong, J. C., Liu, Y., & Zhang, W. (2024). Chimeric antigen receptor-natural killer cell therapy: Current advancements and strategies to overcome challenges. Frontiers in Immunology, 15, 1384039. https://doi.org/10.3389/fimmu.2024.1384039
[13]. Jiang, B., Zhao, L., & Wei, L. (2020). Stroma-targeting therapy in pancreatic cancer: One coin with two sides? Frontiers in Oncology, 10, 576399. https://doi.org/10.3389/fonc.2020.576399
[14]. Li, Q., Lin, H., & Zhou, W. (2019). Abstract A014: Phase I clinical trial with PD-1/MUC1 CAR-pNK92 immunotherapy. Cancer Immunology Research, 7(2_Supplement), A014-A014. https://doi.org/10.1158/2326-6066.CIR-19-A014
[15]. Klichinsky, M., & Larson, S. (2020). Human chimeric antigen receptor macrophages for cancer immunotherapy. Nature Biotechnology, 38(8), 947–953. https://doi.org/10.1038/s41587-020-0509-x
[16]. Sloas, C., Gill, S., & Klichinsky, M. (2021). Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Frontiers in Immunology, 12, 783305. https://doi.org/10.3389/fimmu.2021.783305
[17]. Zhang, W., Wang, T., & Wang, Q. (2019). Chimeric antigen receptor macrophage therapy for breast tumors mediated by targeting the tumor extracellular matrix. British Journal of Cancer, 121(10), 837–845. https://doi.org/10.1038/s41416-019-0495-1
[18]. Li, J., Chen, P., & Ma, W. (2024). The next frontier in immunotherapy: Potential and challenges of CAR-macrophages. Experimental Hematology & Oncology, 13(1), 76. https://doi.org/10.1186/s40164-024-00302-1
[19]. Huang, T., Li, F., & Li, X. (2024). CAR-macrophage: Breaking new ground in cellular immunotherapy. Frontiers in Cell and Developmental Biology, 12, 1464218. https://doi.org/10.3389/fcell.2024.1464218
[20]. Włodarczyk, M., & Pyrzynska, B. (2022). CAR-NK as a rapidly developed and efficient immunotherapeutic strategy against cancer. Cancers, 15(1), 117. https://doi.org/10.3390/cancers15010117
[21]. Schepisi, G., Berardi, R., & Giampieri, R. (2023). The new frontier of immunotherapy: Chimeric antigen receptor T (CAR-T) cell and macrophage (CAR-M) therapy against breast cancer. Cancers, 15(5), 1597. https://doi.org/10.3390/cancers15051597
[22]. Dong, X., Zhang, Y., & Li, L. (2023). Efficacy evaluation of chimeric antigen receptor-modified human peritoneal macrophages in the treatment of gastric cancer. British Journal of Cancer, 129(3), 551–562. https://doi.org/10.1038/s41416-023-02047-6
Cite this article
Dong,J.;Zhang,K. (2025). The Advances of CAR-T, CAR-NK, CAR-M in Solid Tumor Immunotherapy. Theoretical and Natural Science,68,121-130.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 3rd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., & Bray, F. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 71(3), 209–249. https://doi.org/10.3322/caac.21660
[2]. Huo, Y., Zhao, X., Wang, X., Zhang, H., Wang, X., Zhou, L., & Zhang, H. (2023). M1 polarization enhances the antitumor activity of chimeric antigen receptor macrophages in solid tumors. Journal of Translational Medicine, 21(1), 225. https://doi.org/10.1186/s12967-023-04206-z
[3]. Shin, M. H., Lee, H. J., & Kwon, J. (2023). Recent advances in CAR-based solid tumor immunotherapy. Cells, 12(12), 1606. https://doi.org/10.3390/cells12121606
[4]. Alnefaie, A., Albogami, S., Asiri, Y., Ahmad, T., Alotaibi, S. S., Al-Sanea, M. M., & Althobaiti, H. (2022). Chimeric Antigen Receptor T-Cells: An overview of concepts, applications, limitations, and proposed solutions. Frontiers in Bioengineering and Biotechnology, 10, 797440. https://doi.org/10.3389/fbioe.2022.797440
[5]. Straathof, K., Hough, R., & Xie, F. (2020). Antitumor activity without on-target off-tumor toxicity of GD2–chimeric antigen receptor T cells in patients with neuroblastoma. Science Translational Medicine, 12(571), eabd6169. https://doi.org/10.1126/scitranslmed.abd6169
[6]. Junghans, R. P., & Nguyen, L. (2016). Phase I trial of anti‐PSMA designer CAR‐T cells in prostate cancer: Possible role for interacting interleukin 2‐T cell pharmacodynamics as a determinant of clinical response. The Prostate, 76(14), 1257–1270. https://doi.org/10.1002/pros.23212
[7]. Han, J., Zhang, X., & Wang, J. (2015). CAR-engineered NKs targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Scientific Reports, 5, 11483. https://doi.org/10.1038/srep11483
[8]. Wrona, E., Borowiec, M., & Potemski, P. (2021). CAR-NKs in the treatment of solid tumors. International Journal of Molecular Sciences, 22(11), 5899. https://doi.org/10.3390/ijms22115899
[9]. Köylijärvi, M., Koskinen, K., & Jäntti, J. (2020). Translation of small-scale CAR-T cell manufacturing methods to a clinical-scale production platform. Cytotherapy, 22(5), S127–S128. https://doi.org/10.1016/j.jcyt.2020.02.107
[10]. Zhang, L., & Liu, C. (2022). Natural Killer Cell-Based Cancer Immunotherapy: From Bench to Bedside. In Natural Killer Cells: Lessons and Challenges (pp. 25–48). Springer. https://doi.org/10.1007/978-3-030-63271-9_2
[11]. Rossi, F., Pappalardo, S., & Fava, L. (2022). Next generation natural killer cells for cancer immunotherapy. Frontiers in Immunology, 13, 886429. https://doi.org/10.3389/fimmu.2022.886429
[12]. Kong, J. C., Liu, Y., & Zhang, W. (2024). Chimeric antigen receptor-natural killer cell therapy: Current advancements and strategies to overcome challenges. Frontiers in Immunology, 15, 1384039. https://doi.org/10.3389/fimmu.2024.1384039
[13]. Jiang, B., Zhao, L., & Wei, L. (2020). Stroma-targeting therapy in pancreatic cancer: One coin with two sides? Frontiers in Oncology, 10, 576399. https://doi.org/10.3389/fonc.2020.576399
[14]. Li, Q., Lin, H., & Zhou, W. (2019). Abstract A014: Phase I clinical trial with PD-1/MUC1 CAR-pNK92 immunotherapy. Cancer Immunology Research, 7(2_Supplement), A014-A014. https://doi.org/10.1158/2326-6066.CIR-19-A014
[15]. Klichinsky, M., & Larson, S. (2020). Human chimeric antigen receptor macrophages for cancer immunotherapy. Nature Biotechnology, 38(8), 947–953. https://doi.org/10.1038/s41587-020-0509-x
[16]. Sloas, C., Gill, S., & Klichinsky, M. (2021). Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Frontiers in Immunology, 12, 783305. https://doi.org/10.3389/fimmu.2021.783305
[17]. Zhang, W., Wang, T., & Wang, Q. (2019). Chimeric antigen receptor macrophage therapy for breast tumors mediated by targeting the tumor extracellular matrix. British Journal of Cancer, 121(10), 837–845. https://doi.org/10.1038/s41416-019-0495-1
[18]. Li, J., Chen, P., & Ma, W. (2024). The next frontier in immunotherapy: Potential and challenges of CAR-macrophages. Experimental Hematology & Oncology, 13(1), 76. https://doi.org/10.1186/s40164-024-00302-1
[19]. Huang, T., Li, F., & Li, X. (2024). CAR-macrophage: Breaking new ground in cellular immunotherapy. Frontiers in Cell and Developmental Biology, 12, 1464218. https://doi.org/10.3389/fcell.2024.1464218
[20]. Włodarczyk, M., & Pyrzynska, B. (2022). CAR-NK as a rapidly developed and efficient immunotherapeutic strategy against cancer. Cancers, 15(1), 117. https://doi.org/10.3390/cancers15010117
[21]. Schepisi, G., Berardi, R., & Giampieri, R. (2023). The new frontier of immunotherapy: Chimeric antigen receptor T (CAR-T) cell and macrophage (CAR-M) therapy against breast cancer. Cancers, 15(5), 1597. https://doi.org/10.3390/cancers15051597
[22]. Dong, X., Zhang, Y., & Li, L. (2023). Efficacy evaluation of chimeric antigen receptor-modified human peritoneal macrophages in the treatment of gastric cancer. British Journal of Cancer, 129(3), 551–562. https://doi.org/10.1038/s41416-023-02047-6