







1. Introduction
Alzheimer's, an insidious neurodegenerative condition, typically initiates with gradual onset and continually exacerbates over time, accounting for 60–70% of all dementia instances. Memory lapses concerning recent occurrences are frequently the initial manifestation. As the malady progresses, affected individuals may encounter linguistic difficulties, spatial disorientation, which may result in frequent instances of becoming lost, emotional instability, diminished drive, neglect of personal care, and conduct disorders. As the sufferer's health deteriorates, social withdrawal from loved ones and community becomes prevalent. Eventually, the decline encompasses physical capabilities, culminating in mortality. Despite variations in the rate of deterioration, the median survival period post-diagnosis spans approximately three to twelve years.
The origins of Alzheimer's remain a mystery, yet the prevailing theories essentially boil down to a pair of concepts: the amyloid-β (Aβ) pathway and the excessive phosphorylation of the tau protein. According to the Aβ pathway theory, the accumulation of Aβ in the form of inflammatory plaques in the brain leads to the onset of AD by harming the neurons. However, there is no content results in AD clinical treatment for disposing Aβ oligomers. Lately, there has been a surge of interest in the tau protein, as neurotic knots laden with excessively phosphorylated tau are indicative of Alzheimer's disease pathology, and tau plays a pivotal role in the toxic effects of Aβ. Since Aβ oligomers and tau tangles are the prime pathological signs, we want to find out the relationship and interaction between them. In this work, we have analyzed and concluded experiment data. We aspire that this document aids in enhancing the comprehension of the relationship between Aβ and tau in the context of Alzheimer's disease pathogenesis, thereby encouraging additional studies into the interplay mechanics of Aβ and tau.
2. Main body
The amyloid precursor protein (APP) is anchored within the cellular membrane, with one segment residing inside the cell and the other protruding externally. It facilitates the growth and post-injury recuperation of neurons. Being a protein, APP undergoes utilization and eventually degrades, undergoing a recycling process. Typically, it is cleaved by enzymes known as α-secretase and γ-secretase, resulting in a soluble peptide that is easily disposable. However, when the enzyme β-secretase collaborates with γ-secretase, the resulting residual fragment is insoluble and forms a monomer known as Aβ. These adhesive monomers adhere to one another outside neuronal cells, leading to the formation of Aβ plaques. These monomers constitute oligomers (os), and Aβos serve as intermediate stages in the aggregation of Aβ, occurring during both the lag phase and the growth phase. (Figure 1)
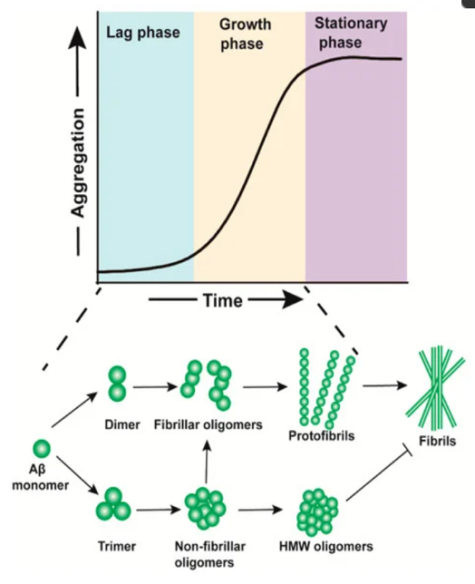
Figure 1: Schematic representation of the process of β-amyloid (Aβ) aggregation
Tau protein basically is a low molecular weight and related to tubulin protein. It mainly exists inside neuron. The role of it is like a transport tool, connecting electrical signal between each neuron. To promote it, there are some modifications on it and the most common one is phosphorate acid. However, it will hyperphosphorylation in AD patient’s neurons. In a comprehensive breakdown, the formation of Aβ plaques exterior to the neuron sets off concurrent internal pathways. This results in the triggering of kinase activity, which is an enzymatic process that attaches phosphate groups to the tau protein. The precise locations wherephosphorate acid are situated within the N-terminal segment (including Ser46, Thr123, Ser198, Ser199, Ser202, Ser208, Ser210, Thr212, Ser214, Thr217, Thr231, and Ser235), the repeat domain (Ser262 and Ser356), and the C-terminal area (Ser396, Ser400, Thr403, Ser404, Ser409, Ser412, Ser413, and Ser422), as depicted in Figure 1. Multiple enzymes are responsible for the hyperphosphorylation of these sites, among them A-kinase, C-kinase, cyclin-dependent kinase-5 (CDK-5), CaM kinase II, glycogen synthase kinase-3β (GSK-3β), and MAPKs, as outlined in Table 1. Consequently, the tau protein undergoes a structural transformation due to its detachment from microtubules (MTs) and the subsequent formation of intracellular neurofibrillary tangles. This disrupts the tau protein's role in supporting the MTs within the cytoskeleton, leading to aggregation with other tau proteins and forming tangled structures known as neurofibrillary tangles. Neurons affected by these tangles and malfunctioning microtubules suffer impaired signaling and may eventually undergo apoptosis, or controlled cell death. To encapsulate, the Aβ protein acts as a catalyst, exacerbating the hyperphosphorylation of the tau protein.
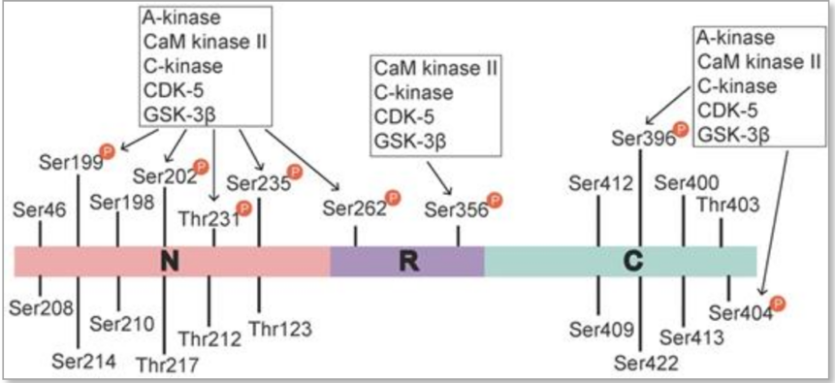
Figure 2: Tau phosphorylation sites and associated kinases
Table 1: Sites on tau phosphorylated by different kinases
Phosphorylation stage | Kinase | Phosphorylation sites | Whether Aβ is involved |
Prephosphorylation | A-kinase | Ser262,Ser294,Ser305, Ser324,Ser356 | No |
C-kinase | Ser305 | No | |
CaM kinase | Ser416/Ser262 | No | |
CDK-5 | Ser195,Ser202,Thr231, Ser235,Ser396,Ser404 | Yes | |
Phosphorylation | GSK-3β | Ser199*,Ser202,Thr231*, Ser235,Ser262,Ser396*, Ser404* | Yes |
MAPK | Thr181,Ser202*,Thr205*, Ser396*,Ser404*,Ser422, Ser199*,Thr50* | Yes | |
Prephosphorylation+Phosphorylation | A-kinase+GSK-3β | Ser199*,Ser202*,Thr231, Ser235,Ser262,Ser396*, Ser404d* | Yes |
CaM kinase+GSK-3β | Ser199*,Ser202*,Thr231*, Ser235*,Ser262*,Ser396*, Ser404* | Yes | |
CDK-5+GSK-3β | Ser199,Ser202,Thr231*, Ser235,Ser262,Ser396, Ser404d | Yes |
Aβ plaques accelerate neurotic plaque tau aggregation and propagation. A new hypothesis states that the medium of it is τ protein. This protein also can be phosphorylated. The propagation of Aβ and phosphorylated-τ pathologies across the brain follows a structured, stepwise progression. Notably, an aggregation of phosphorylated-τ is evident within the locus coeruleus, raphenuclei, substantia nigra, the dorsal nucleus of the vagus nerve, and the basal nucleus of Meynert. Subsequent to their maturation, Aβ deposits become visible in distinct brain areas. Latest findings depict that the cellular prion protein (PrPC) acts as a receptor for harmful Aβ variants and -synuclein aggregates. PrPC is present in the neuropil and has been identified within amyloid deposits and neurons in Alzheimer's disease patients. Concentrated in postsynaptic densities, PrPC triggers the activation of Fyn kinase. This activated kinase phosphorylates the GluN2B subunit of NMDA receptors and engages with the phosphorylation of tau. As Aβ aggregates prompt PrPC-Fyn mediated phosphorylation of tau, PrPC could be involved in the interaction between Aβ and phosphorylated-τ either directly or indirectly. Synaptic terminals release soluble phosphorylated-τ. P-τ can either form inside the cell or outside the cell. When it form ouside the cell, there is opputunity forsoluble p-τ combine with PrPC. Aβ is also combines eith PrPC and stimulates Fyn, through pyk2 related tau phosphorylation increases degree of p-τ. Neurons lacking tau demonstrate an immunity to the degenerative effects caused by either synthetic or human origin Aβ peptides, whereas an increase in tau expression intensifies the detrimental impact of Aβ. In this context, the interaction between Aβ and PrPC, the activation of Fyn, and the phosphorylation of the tau protein could offer different perspectives on the observation that Aβ deposits expedite the spread of tau phosphorylation through a PrPC-associated pathway, potentially promoting tau accumulation in brain regions with Aβ buildup. Consequently, phosphorylated tau might serve as an additional protein involved in the interplay between Aβ and tau. The interconnections observed at the site of damage further illustrate the complex network of direct and indirect relationships between Aβ deposits and tau clumping.
This study aims to gauge the impact of Aβ and tau protein clumping. Utilizing tau HEK293T cells equipped with a biosensor, these cells carry the P301S mutated tau repeat domain (RD) fused with either yellow or cyan fluorescent proteins. As tau proteins clump together within these cells, the yellow and cyan fluorescent proteins create a FRET pair, enabling the spectral quantification of the aggregate clusters. The cells are initially exposed to various Aβ species (recently prepared, oligomeric, and fibrillar) via lipofectamine, followed by the introduction of fibrillar tau RD seeds 24 hours into the process. Post this interval, fluorescent microscopy is employed to capture imagery. In the absence of tau seed transduction, the biosensor cells exhibit a diffused fluorescent signal, suggesting the baseline expression of non-clumped, native tau. When the cells are seeded with tau RD at a concentration of 10 nM, they reveal intracellular tau aggregates as bright fluorescent spots. An increase in the concentration of Aβ oligomers results in a marked rise in the visibility of these fluorescent puncta. The findings indicate that the stimulation of tau aggregation by Aβ oligomers is proportional to their concentration, with the influence of newly prepared and fibrillar Aβ being significantly subtler. To further delineate these varying influences, the quantity of tau fibril seeds in the aggregation assay was manipulated.
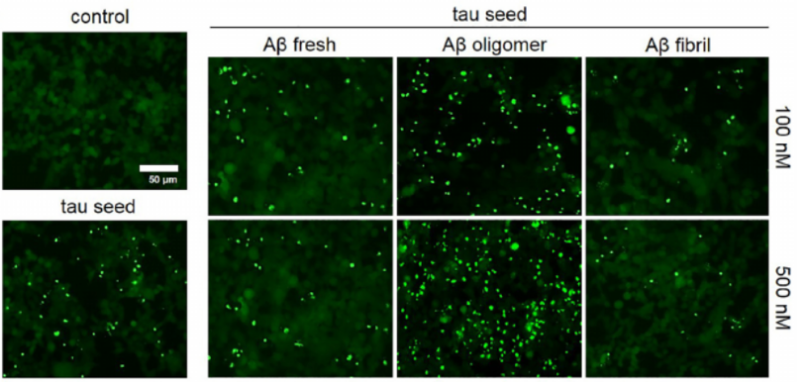
Figure 3: Fluorescent microscope images of seeded biosensor cells
3. Conclusion
In summary, Aβ and tau proteins are pivotal in Alzheimer's disease, contributing to the characteristic observed in the brains of deceased patients, namely amyloid deposits and neurofibrillary tangles. Despite their significant contribution to the disease's onset, monotherapies directed at Aβ or tau have failed to yield satisfactory therapeutic outcomes in clinical trials. Thus, people should pay concentration on the interaction or relationship research and make detailed and logical conclusion. Now we know Beta-amyloid hastens the phosphorylation process of the tau protein, while it concurrently disrupts the aggregation of tau into oligomers. The detrimental impact of beta-amyloid is contingent upon its interaction with tau, as both proteins collaborate to compromise mitochondrial function. Investigations both in the laboratory and in clinical settings have been carried out, affirming the theory that the synergy between beta-amyloid and tau magnifies the deleterious impact of both. Attempts have been undertaken to disrupt the association between beta-amyloid and the tau protein, including research into inhibitors of GSK-3β and CDK-5. Nevertheless, the therapeutic effectiveness in a clinical context remains undetermined, and the inquiry into the relationship between beta-amyloid and tau persists. Given that Alzheimer's disease is a multifaceted condition, likely stemming from a confluence of genomic, epigenetic, atomic, and environmental factors, it is imperative to concentrate not solely on medicinal treatments but also on the intricate biopsychosocial dimensions of managing AD, such as devising psychological therapies. Additionally, the pursuit of novel pharmacological agents capable of addressing AD through diverse pathways warrants further exploration.
References
[1]. Zhang, H., Wei, W., Zhao, M., Ma, L., Jiang, X., Pei, H., Cao, Y., Li, H. (2021). Interaction between Aβ and Tau in the Pathogenesis of Alzheimer's Disease. International Journal of Biological Sciences, 17(9), 2181-2192. https://doi.org/10.7150/ijbs
[2]. Rodriguez-Vieitez, E., Montal, V., Sepulcre, J. et al. Association of cortical microstructure with amyloid-β and tau: impact on cognitive decline, neurodegeneration, and clinical progression in older adults. Mol Psychiatry 26, 7813–7822 (2021). https://doi.org/10.1038/s41380-021-01290-z
[3]. Shin, W.S., Di, J., Cao, Q. et al. Amyloid β-protein oligomers promote the uptake of tau fibril seeds potentiating intracellular tau aggregation. Alz Res Therapy 11, 86 (2019). https://doi.org/10.1186/s13195-019-0541-9
[4]. Cai, Y., Du, J., Li, A. et al. Initial levels of β-amyloid and tau deposition have distinct effects on longitudinal tau accumulation in Alzheimer’s disease. Alz Res Therapy 15, 30 (2023). https://doi.org/10.1186/s13195-023-01178-w
[5]. Wang C, Holtzman DM. Bidirectional relationship between sleep and Alzheimer's disease: role of amyloid, tau, and other factors. Neuropsychopharmacology. 2020 Jan;45(1):104-120.
[6]. Karran E, De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov. 2022 Apr;21(4):306-318
[7]. Chen Y, Yu Y. Tau and neuroinflammation in Alzheimer's disease: interplay mechanisms and clinical translation. J Neuroinflammation. 2023 Jul 14;20(1):165.
[8]. Doré V, Krishnadas N, Bourgeat P, Huang K, Li S, Burnham S, Masters CL, Fripp J, Villemagne VL, Rowe CC. Relationship between amyloid and tau levels and its impact on tau spreading. Eur J Nucl Med Mol Imaging. 2021 Jul;48(7):2225-2232.
[9]. Zhao Y, Liu B, Wang J, Xu L, Yu S, Fu J, Yan X, Su J. Aβ and Tau Regulate Microglia Metabolism via Exosomes in Alzheimer's Disease. Biomedicines. 2022 Jul 27;10(8):1800.
[10]. Int. J. Mol. Sci. 2020, 21(12), 4477; https://doi.org/10.3390/ijms21124477
Cite this article
Li,S. (2025). Relationship Between Amyloid-beta Deposition and Tau Deposition in Alzheimer's Disease. Theoretical and Natural Science,116,8-12.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 3rd International Conference on Modern Medicine and Global Health
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Zhang, H., Wei, W., Zhao, M., Ma, L., Jiang, X., Pei, H., Cao, Y., Li, H. (2021). Interaction between Aβ and Tau in the Pathogenesis of Alzheimer's Disease. International Journal of Biological Sciences, 17(9), 2181-2192. https://doi.org/10.7150/ijbs
[2]. Rodriguez-Vieitez, E., Montal, V., Sepulcre, J. et al. Association of cortical microstructure with amyloid-β and tau: impact on cognitive decline, neurodegeneration, and clinical progression in older adults. Mol Psychiatry 26, 7813–7822 (2021). https://doi.org/10.1038/s41380-021-01290-z
[3]. Shin, W.S., Di, J., Cao, Q. et al. Amyloid β-protein oligomers promote the uptake of tau fibril seeds potentiating intracellular tau aggregation. Alz Res Therapy 11, 86 (2019). https://doi.org/10.1186/s13195-019-0541-9
[4]. Cai, Y., Du, J., Li, A. et al. Initial levels of β-amyloid and tau deposition have distinct effects on longitudinal tau accumulation in Alzheimer’s disease. Alz Res Therapy 15, 30 (2023). https://doi.org/10.1186/s13195-023-01178-w
[5]. Wang C, Holtzman DM. Bidirectional relationship between sleep and Alzheimer's disease: role of amyloid, tau, and other factors. Neuropsychopharmacology. 2020 Jan;45(1):104-120.
[6]. Karran E, De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov. 2022 Apr;21(4):306-318
[7]. Chen Y, Yu Y. Tau and neuroinflammation in Alzheimer's disease: interplay mechanisms and clinical translation. J Neuroinflammation. 2023 Jul 14;20(1):165.
[8]. Doré V, Krishnadas N, Bourgeat P, Huang K, Li S, Burnham S, Masters CL, Fripp J, Villemagne VL, Rowe CC. Relationship between amyloid and tau levels and its impact on tau spreading. Eur J Nucl Med Mol Imaging. 2021 Jul;48(7):2225-2232.
[9]. Zhao Y, Liu B, Wang J, Xu L, Yu S, Fu J, Yan X, Su J. Aβ and Tau Regulate Microglia Metabolism via Exosomes in Alzheimer's Disease. Biomedicines. 2022 Jul 27;10(8):1800.
[10]. Int. J. Mol. Sci. 2020, 21(12), 4477; https://doi.org/10.3390/ijms21124477