







1. Introduction
Since the first separation of exaltolide from Kerschbaum in 1927, macrolide compounds have entered the human eye. The most prominent feature of these compounds is that they can be used as antibiotics, such as erythromycin, azithromycin, etc., whose biological activity is highly dependent on the unique conformation of their macrocyclic structure. Therefore, in the process of research and synthesis of these important drugs, macrocyclic esterification has turned to be a topic worthy of further discussion.
As a key organic synthesis method, macrocyclic esterification has been extensively studied for decades. To address challenges like intermolecular esterification reaction, mid-ring strain, and the inertness of alcohol and acid substrates, designing appropriate synthesis strategies and activation reagents based on the basic principles of organic chemistry has become central to the research area.
At present, the development direction of traditional synthesis strategy lies in how to activate carboxyl or hydroxyl groups of substrates specifically and efficiently, and develop more favorable activating reagents based on this, or adopt more efficient reaction methods to improve the yield of target products. In recent years, some new reaction ideas started from the perspective of substrate design, and the specific redox reaction was realized by catalysts to indirectly construct macrolides, which broadened the synthesis strategy of such reactions. In a word, the development of these strategies has provided fundamental help for the synthesis of macrolides and biomedical research, and it is also a great progress in organic methodology.
This article focuses on the specific development of macrolide technique in the contemporary scientific research environment. This research is not only expected to drive technological progress in the synthesis of macrolide drugs but also to provide valuable insights and references for researchers in related fields.
2. Traditional synthesis strategies
2.1. Basic reaction mode of carboxyl activation
The essence of macrocyclic esterification is to remove a molecule of water from the carboxyl and hydroxyl groups and promote the formation of lactone bonds, and the activation mechanism of carboxyl group is to convert the hydroxyl group in the carboxyl group into a suitable leaving group, and enhance the electrophilicity of the carbonyl group in the carboxyl group under the catalysis of Lewis base, so that the hydroxyl group can better combine with the carbonyl group.
The reagents used to complete these classical carboxyl activations include acyl halides, anhydrides, halopyridine cations, carbodiimide reagents and organophosphorus reagents, which have derived many name reactions such as Yamaguchi reaction, Mukaiyama reaction, Shiina reaction, etc. Several classic methods and extension ideas will be briefly explained.
2.2. Yamaguchi esterification
Yamaguchi esterification was first reported in 1979, and its core activating reagent is 2,4,6-trichlorobenzoyl chloride (TCBC) [1-2]. This type of acyl chloride has strong activity due to the substitution of chlorine atoms on the benzene ring. Therefore, it can react with carboxyl group quickly to produce an activated intermediate mixed anhydride under the action of triethylamine. Finally, the product of macrolide is obtained under the catalysis of 4-N,N-dimethylaminopyridine (DMAP), a commonly used nucleophilic catalyst, and the TCBC part finally leaves the reaction system in the form of carboxylate anion [3].
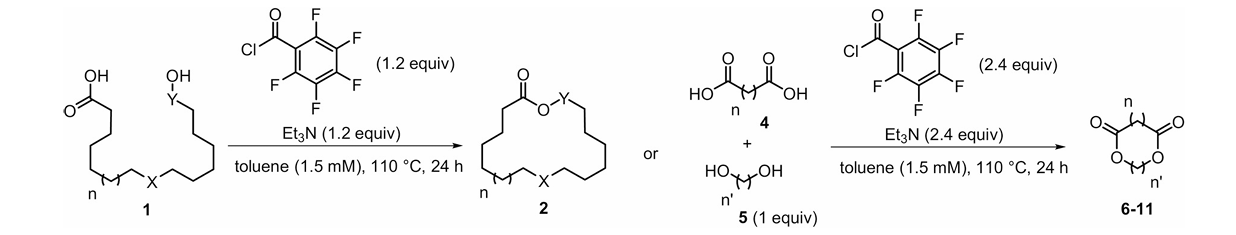
Since these reactions share similar mechanisms, the optimization mainly focus on the design and replacement of reagent substituents. For example, in 2021, Ilaria Ciofini and David Lebœuf et al. explored the impact of the conversion of trichlorophenyl to pentafluorophenyl in TCBC on macrocyclic esterification. As shown in Fig.1, they explored the intermolecular condensation of various substrates with two macrolide forms — hydroxyl carboxylic acid cyclization and diols-diacid condensation. it is finally determined that pentafluorobenzoyl chloride can greatly improve the yield of macrolide and is compatible with diverse functional groups. Density Functional Theory (DFT) calculations revealed that pentafluorobenzoyl could pre-organize the substrate through non-covalent interactions (including hydrogen bonds between fluorine and oxygen atoms and lp-π phase interactions between electron-deficient benzene ring and hydroxyl end) after the formation of mixed anhydride intermediate, thus favoring intramolecular condensation over intermolecular oligomerization. Additionally, pentafluorophenyl group reagent is cheap, and the by-product pentafluorobenzoic acid, is highly water-soluble, facilitating its removal from the organic phase. In conclusion, the replacement of such substituents can better overcome the intermolecular side reactions in the macrocyclic esterification reaction and greatly improve the reaction yield [4].
2.3. Mukaiyama esterification
Mukaiyama esterification initiated in 1976 is also a typical case of carboxyl activation using pyridinium halide (Mukaiyama salt) as the activating reagent [5-7]. The unique electrophilic nature of the cation, with its chlorine-substituted pyridine ring and positively charged nitrogen atom, enables it to react with the carboxyl group's oxygen, forming a stable N-methylpyridinone by-product that drives the reaction forward
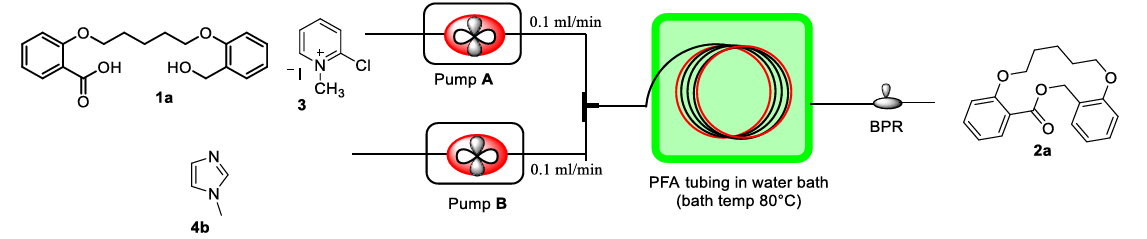
Dashrat Vishambar Sutar et al. explored enhancing the Mukaiyama reaction through continuous flow under high dilution conditions. This well-established technology is often used in the large-scale production of intermediates and pharmaceutical drugs. Different reaction bottoms are dynamically transported to the reactor through flexible pumps, so that the reaction system can accurately control the high dilution conditions of the esterification process in the macrocycle. As shown in Fig.2, in the specific work that has been published, the hydroxy acid substrate with the Mukaiyama reagent and the base 1-methylimidazole (MIM) were respectively put into two independent pumps and injected into the perfluoroalkoxy alkane (PFA) tubular reactor at a certain flow rate. The target macrolide product was finally obtained under the condition of water bath heating at 80 ℃. This method has been successfully applied to the synthesis of more than ten kinds of macrolides with its short reaction time, significantly high yield and wide range of application [8].
3. New synthesis strategies
In recent years, the ways to realize the esterification in the macrocycle are not limited to the idea of activating the carboxyl and hydroxyl groups, but gradually put the focus of ring forming into the idea of designing substrates and then using new catalysts, such as the oxidative esterification of aldehydes catalyzed by N-heterocyclic carbenes (NHC) and the catalytic dehydrogenative intramolecular macrolactonization process involving metal catalysts for diol substrates [8].
3.1. NHC strategies
NHC catalyst, a widely-used polarity-reversal catalyst, undergoes a nucleophilic reaction with aldehydes. This process converts the hydrogen atom on the aldehyde group into acid hydrogen atom, thus generating the enol intermediate after polarity reversal. If there are single electron oxidants in the system, the intermediate can be oxidized to an activated acyl intermediate. Subsequently, the alcohol hydroxyl at the other end can be nucleophilic substituted with the activated acyl intermediate, yielding macrolides upon catalyst departure. Additionally, because NHC catalyst can be modified to be chiral, this kind of reaction can be used to construct macrolides with aromatic ring-hindered isomerization ring system.
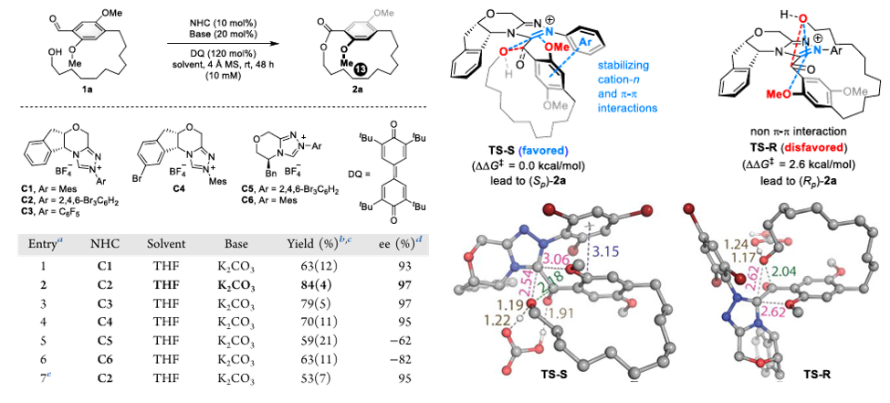
In 2024, Chenyang Li et al. and Xingxing Wu et al. reported the related work of NHC-catalyzed macrocyclic esterification of aldehydes. Li et al. took the substrate containing dimethoxy-substituted benzene as a case, and relied on sufficient DFT theory to calculate the evidence, focusing on the cation-n and π-π interaction between NHC catalyst substituted by aromatic groups and the substrate. As depicted in Fig.3, the cationic ring system of NHC attracts cation and lone pair with methoxy group in the substrate and hydroxyl group that will undergo nucleophilic reaction. Meanwhile, the aromatic ring (2,4,6 - tribromophenyl group) in the preselected catalyst interacts π-π with electron-rich benzene ring in the substrate, thus generating the most suitable stable transition state, and finally reacting to generate the chiral hindered isomerization esterification product. This reaction methodology has been successfully applied to the synthesis of various products [9]. For Wu et al.’s work, they used bromonaphthalene ring substrate, and by adding suitable hydrogen bond donors (HBD) into the system, the thiourea group of the catalyst HBD-3 can interact with the hydroxyl group in the substrate and the nitrogen heterocyclic ring in the NHC catalyst for additional hydrogen bonds, which also stabilized the transition state of the product and finally obtained the best yield. After obtaining the product, it can be appropriately modified by Suzuki reaction and other coupling methods to expand its application in synthesis [10].
3.2. Transition metal catalysts strategies
Transition metal catalytic reaction shows amazing application in organic synthesis with its unique reaction mode. Because the metal center usually activates functional groups or chemical bonds in the form of oxidation state change, the corresponding substrate can be designed to achieve a redox synthesis of macrolides by using this property of metal catalysis. Among them, the most classic strategy is the catalytic dehydrogenation and lactonization of diol substrate, and hydrogen-loving metals such as ruthenium and rhodium are adopted as the catalytic core to achieve the purpose of adsorbing hydrogen atoms.
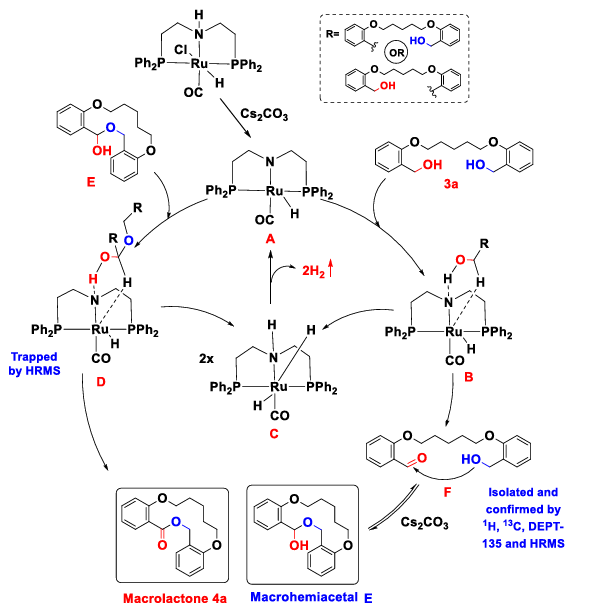
In 2022, Boopathy Gnanaprakasam et al. used Ru-MACHO as a ruthenium-based catalyst to successfully realize the catalytic dehydrogenation and lactonization of dibenzyl alcohol system. As shown in Fig.4, cesium carbonate was used as the base to open the active site of ruthenium center, and one-step catalytic dehydrogenation was carried out to produce aldehyde-alcohol intermediates through the hydrogen bond between imino group in the ligand and hydroxyl group in the substrate. After the aldol substrate is condensed to form hemiacetal, hemiacetal is further dehydrogenated, and finally macrolide is generated. The system only needs cesium carbonate as alkali, and the by-product is only hydrogen, which has high application value [11].
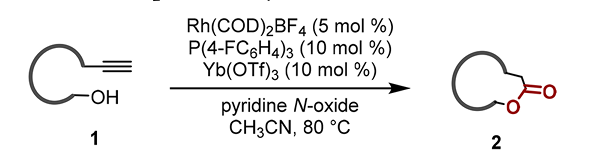
Innovative methods leveraging uncommon substrates and transition metal catalysis have also emerged. A prime example is the 2018 work by Lijin Xu et al. on alkynyl alcohol oxidative cyclization. The key intermediate of metal carbene was obtained by using the affinity between alkynyl and rhodium catalyst after hydrogen migration. Subsequently, the rhodium carbene intermediate was further converted into ketene intermediate by nucleophilic oxidation of pyridine oxide, which is an activated intermediate with high activity but difficult to generate. The substrate's hydroxyl group subsequently combined with the ketene, completing the macrocyclic esterification under ytterbium trifluoroacetate's Lewis acid action. This reaction can be carried out smoothly under the condition of high concentration and no protective group, and has a wide range of substrate applications, according to Fig.5. It is a successful case of metal-catalyzed macrocyclic esterification [12-13].
4. Conclusion
Up to now, there have been many kinds of methodologies for macrocyclic esterification, among which the traditional activation route is still the main synthesis method, and various new synthesis methods are emerging one after another. Generally speaking, because the method of carboxyl activation has a long history and has been updated and iterated, making them widely applicable in many related total syntheses. In contrast, due to the difficulty in designing the catalytic system and the particularity of the substrate, the new synthesis strategy based on abnormal substrates has not been widely used in total synthesis. For instance, non-activation strategies are rarely employed in recent macrolide total syntheses.
There have been various outstanding achievements in the extension and expansion of the reaction these years, but the improvements in traditional synthetic ideas often focus on optimizing existing reagent substituents and reaction conditions, and there is a lack in finding new activators. The new synthetic idea will inevitably affect its work because of the limitation of substrates and catalysts, and the expansion of substrates mostly lies in changing carboxyl groups but lacks the work of changing hydroxyl groups, which may become another possible development idea.
This study acknowledges several limitations. Firstly, the exploration of new activators and catalysts is still in its nascent stages, with much work remaining to be done. Secondly, while the study provides a comprehensive overview of current methodologies, it may not fully capture the nuances of emerging techniques. Future research direction in this field is likely to focus on the application of new catalysts in macrocyclic esterification and the further search for new activators, further advancing the field of macrolide chemistry.
References
[1]. Inanaga, J., Hirata, K., Saeki, H., Katsuki, T., & Yamaguchi, M. (1979). Bulletin of the Chemical Society of Japan, 52, 1989-1993.
[2]. Kawanami, Y., Dainobu, Y., Inanaga, J., Katsuki, T., & Yamaguchi, M. (1981). Bulletin of the Chemical Society of Japan, 54, 943-944.
[3]. Majhi, S. (2021). Applications of Yamaguchi Method to Esterification and Macrolactonization in Total Synthesis of Bioactive Natural Products. ChemistrySelect, 6, 4178–4206.
[4]. Force, G., Perfetto, A., Mayer, R. J., Ciofini, I., & Lebœuf, D. (2021). Macrolactonization Reactions Driven by a Pentafluorobenzoyl Group. Angewandte Chemie International Edition, 60, 19843–19851.
[5]. Mukaiyama, T., Usui, M., Shimada, E., & Saigo, K. (1975). Chemical Letters, 1045-1048.
[6]. Hojo, K., Kobayashi, S., Soai, K., Ikeda, S., & Mukaiyama, T. (1977). Chemical Letters, 635-636.
[7]. Mukaiyama, T. (1979). Angewandte Chemie International Edition, 18, 707-708.
[8]. Sutar, D. V., Sarang, N. U., Jamdade, A. B., & Gnanaprakasam, B. (2023). Continuous Flow Inter- and Intramolecular Macrolactonization under High Dilution Conditions. Journal of Organic Chemistry, 88, 3740−3759.
[9]. Wang, J., Wang, M., Wen, Y., Teng, P., Li, C., & Zhao, C. (2024). N-Heterocyclic Carbene-Catalyzed Highly Enantioselective Macrolactonization to Access Planar-Chiral Macrocycles. Organic Letters, 26, 1040−1045.
[10]. Lv, X., Su, F., Long, H., Lu, F., Zeng, Y., Liao, M., Che, F., Wu, X., & Chi, Y. R. (2024). Carbene organic catalytic planar enantioselective macrolactonization. Nature Communications, 15(1), 958.
[11]. Jamdade, A. B., Sutar, D. V., & Gnanaprakasam, B. (2022). Dehydrogenative Intramolecular Macrolactonization of Dihydroxy Compounds Using Ru-MACHO Catalyst. Organic Letters, 24, 4394−4398.
[12]. Zhang, W.-W., Gao, T.-T., Xu, L.-J., & Li, B.-J. (2018). Macrolactonization of Alkynyl Alcohol through Rh(I)/Yb(III) Catalysis. Organic Letters, 20, 6534−6538.
[13]. Zhang, W.-W., Gao, T.-T., Xu, L.-J., & Li, B.-J. (2019). Correction to “Macrolactonization of Alkynyl Alcohol through Rh(I)/Yb(III) Catalysis”. Organic Letters, 21, 10170−10170.
Cite this article
Wang,T. (2025). Macrocyclic Esterification: A Review of Synthetic Methodologies and Their Extensions. Theoretical and Natural Science,119,143-148.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of ICEGEE 2025 Symposium: Sensor Technology and Multimodal Data Analysis 2025
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Inanaga, J., Hirata, K., Saeki, H., Katsuki, T., & Yamaguchi, M. (1979). Bulletin of the Chemical Society of Japan, 52, 1989-1993.
[2]. Kawanami, Y., Dainobu, Y., Inanaga, J., Katsuki, T., & Yamaguchi, M. (1981). Bulletin of the Chemical Society of Japan, 54, 943-944.
[3]. Majhi, S. (2021). Applications of Yamaguchi Method to Esterification and Macrolactonization in Total Synthesis of Bioactive Natural Products. ChemistrySelect, 6, 4178–4206.
[4]. Force, G., Perfetto, A., Mayer, R. J., Ciofini, I., & Lebœuf, D. (2021). Macrolactonization Reactions Driven by a Pentafluorobenzoyl Group. Angewandte Chemie International Edition, 60, 19843–19851.
[5]. Mukaiyama, T., Usui, M., Shimada, E., & Saigo, K. (1975). Chemical Letters, 1045-1048.
[6]. Hojo, K., Kobayashi, S., Soai, K., Ikeda, S., & Mukaiyama, T. (1977). Chemical Letters, 635-636.
[7]. Mukaiyama, T. (1979). Angewandte Chemie International Edition, 18, 707-708.
[8]. Sutar, D. V., Sarang, N. U., Jamdade, A. B., & Gnanaprakasam, B. (2023). Continuous Flow Inter- and Intramolecular Macrolactonization under High Dilution Conditions. Journal of Organic Chemistry, 88, 3740−3759.
[9]. Wang, J., Wang, M., Wen, Y., Teng, P., Li, C., & Zhao, C. (2024). N-Heterocyclic Carbene-Catalyzed Highly Enantioselective Macrolactonization to Access Planar-Chiral Macrocycles. Organic Letters, 26, 1040−1045.
[10]. Lv, X., Su, F., Long, H., Lu, F., Zeng, Y., Liao, M., Che, F., Wu, X., & Chi, Y. R. (2024). Carbene organic catalytic planar enantioselective macrolactonization. Nature Communications, 15(1), 958.
[11]. Jamdade, A. B., Sutar, D. V., & Gnanaprakasam, B. (2022). Dehydrogenative Intramolecular Macrolactonization of Dihydroxy Compounds Using Ru-MACHO Catalyst. Organic Letters, 24, 4394−4398.
[12]. Zhang, W.-W., Gao, T.-T., Xu, L.-J., & Li, B.-J. (2018). Macrolactonization of Alkynyl Alcohol through Rh(I)/Yb(III) Catalysis. Organic Letters, 20, 6534−6538.
[13]. Zhang, W.-W., Gao, T.-T., Xu, L.-J., & Li, B.-J. (2019). Correction to “Macrolactonization of Alkynyl Alcohol through Rh(I)/Yb(III) Catalysis”. Organic Letters, 21, 10170−10170.