







1. Introduction
Alzheimer's disease (AD), a neurodegenerative disease, is the most common cause of dementia worldwide [1]. It is characterised by beta-amyloid(Aβ) deposition and abnormal phosphorylation of tau protein. So far, over 55 million AD patients worldwide, projected to reach 139 million by 2050 [2]. Conventional treatment regimens, often involving cholinesterase inhibitors and N-methyl-D-aspartate (NMDA) antagonists, have been ineffective in slowing progression [3]. Reviewing the pathology of AD, Aβ deposition has been seen 15-20 years earlier than clinical symptoms and is also a key target for AD disease-modifying therapy (DMT) [4]. In 2023, Lecanemab became the first DMT drug fully approved by the U.S. Food and Drug Administration (FDA) for the treatment of AD [5].
The development of Lecanemab marks a breakthrough in the field of anti-Aβ mAb therapy. Developed in 2014 by Eisai (Japan) in collaboration with Biogen (USA), the drug was designed from the concept of precise targeting of Aβ pathological subtypes, unlike earlier antibodies (e.g., Bapineuzumab) that pan-bind to Aβ monomers. Lecanemab works by specifically recognising soluble Aβ protofibrils, a conformation that is more synaptotoxic [6]. Studies before clinical trials further validated the mechanism: in a transgenic mouse model expressing the human Amyloid Precursor Protein (APP) gene, Lecanemab treatment for 12 weeks resulted in a more than 50% reduction in amyloid plaque load in the brain and did not trigger significant vasogenic oedema [7]. This result not only supports the feasibility of Aβ protofibrils as a therapeutic target but also lays the foundation for subsequent human trials. The clinical translation of Lecanemab followed a rigorous stepwise validation pathway. This data not only confirms the efficacy of the Phase II study, but for the first time in a Phase III trial, both statistical and clinical significance were achieved, propelling Lecanemab to full FDA approval in July 2023 [8]. Despite being an anti-Aβ mAb, Lecanemab has a significantly different action profile and risk-benefit profile than its counterparts. Compared to Donanemab, the latter achieves more rapid plaque clearance by targeting the pyroglutamated Aβ plaque core (40% of patients achieve PET-negativity within 6 months), but its ARIA-E incidence is as high as 26.6%, which is significantly higher than Lecanemab's 12.6% [9]. Another controversial drug, Aducanumab, was granted accelerated approval by the FDA in 2021. Still, significant heterogeneity in the results of its phase III trial and lower improvement in cognitive endpoints (CDR-SB difference -0.39, p=0.012) than that of Lecanemab led to the rejection of its marketing application by the European medicines agency (EMA) [10]. Comparison of these drugs highlights the advantages of Lecanemab in terms of efficacy-safety balance, making it currently the only disease-modifying therapy to have gained widespread international acceptance.
The primary objective of this paper is to systematically review the mechanism of action, clinical evidence, and safety profile of Lecanemab, analyze the impact of anti-Aβ monotherapy on AD clinical practice, and explore the future potential of combination therapies (e.g., Aβ+Tau dual-targeting). To provide clinicians with a reference for evidence-based medication decisions, to reveal the prospects of biomarkers in precision therapy, and to promote the transition of AD from symptom management to disease-modifying therapy.
2. The core pathways of AD pathogenesis
2.1. The Aβ cascade hypothesis: from oligomers to plaque deposition
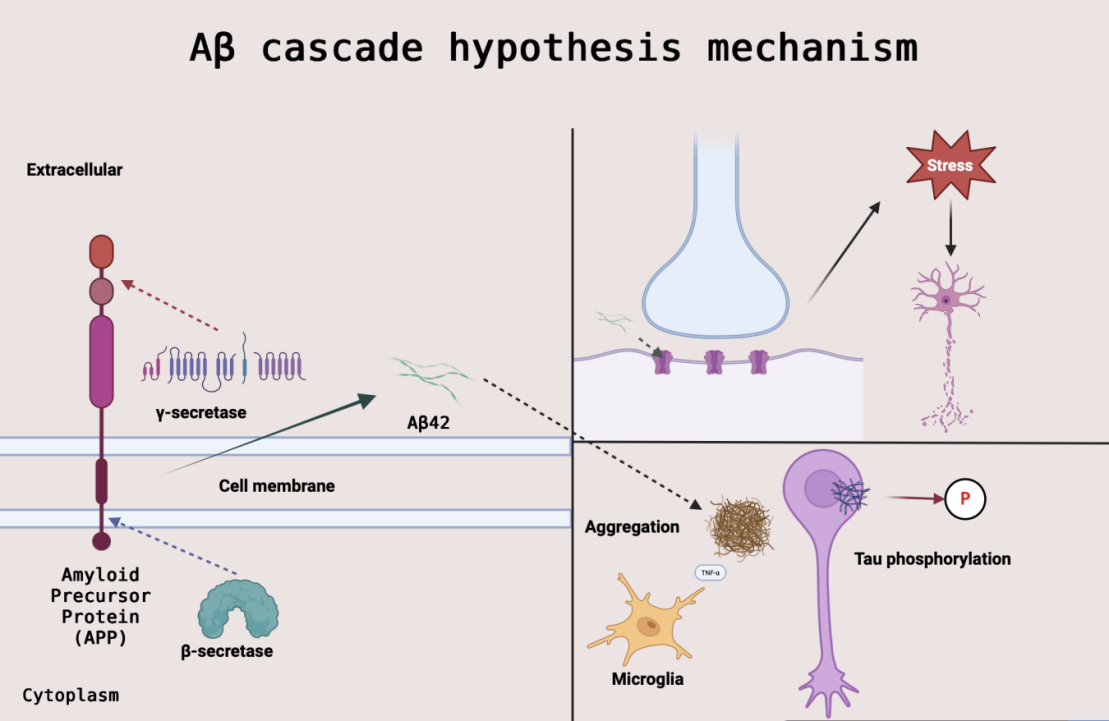
The Aβ cascade hypothesis is the central framework for the pathomechanism of AD and is centered on the idea that aberrant metabolism of Aβ. The Aβ hypothesis was first systematically elaborated by Hardy and Higgins in 1992 [11]. This hypothesis states that APP is abnormally sheared by β-secretase and γ-secretase to generate neurotoxic Aβ oligomers. These oligomers preferentially attack the postsynaptic membrane, directly contributing to synaptic dysfunction and early cognitive decline by interfering with NMDA receptor function and inducing oxidative stress [12].
However, clinical studies have found that amyloid plaque load is not fully consistent with the degree of cognitive decline, and transgenic mouse models (e.g., APP/PS1 mice) have shown that Aβ overexpression induces plaque deposition and neuronal damage [13]. This paradox suggests that soluble Aβ oligomers (rather than plaques themselves) may be the more critical form of toxicity. For example, the failure of clinical trials of the anti-Aβ drug Bapineuzumab was attributed in part to its removal of plaque only and not targeting oligomers [12].
Current research emphasizes the dynamic network effects of Aβ pathology: oligomers drive tau pathology spreading and neuroinflammation through activation of tau phosphorylated kinases (e.g., GSK-3β) and pro-inflammatory signaling pathways [12]. This mechanism provides a theoretical rationale for drugs such as Lecanemab that target soluble Aβ protofibrils (protofibrils) - potentially blocking downstream pathological cascades through early removal of oligomers [13]. The Aβ cascade hypothesis mechanism is illustrated in Figure 1.
2.2. Tau protein pathology and neuro progenitor fiber tangles (NFTs)
Tau proteins maintain axonal transport function by stabilising neuronal microtubules in the physiological state, but their aberrant phosphorylation (e.g., p-tau181 and p-tau217 sites) leads to dissociation from microtubules and aggregation into NFTs, which are a core pathological hallmark of AD [14]. Notably, the spread of Tau pathology follows the Braak staging model: from the internal olfactory cortex (Braak stages I-II) through the hippocampus (stages III-IV) to the neocortex (stages V-VI), with progression directly correlating with the degree of cognitive decline [15].
Synergistic mechanism with Aβ: Aβ oligomers promotes Tau phosphorylation through activation of GSK-3β kinase, while accumulation of NFTs further exacerbates synaptic loss, creating an 'Aβ-Tau pathological positive feedback loop’. This interaction is manifested in the clinic by the fact that elevated plasma p-tau217 (a marker of Tau pathology) precedes Aβ-positive imaging, suggesting that Tau may be a downstream event of Aβ, and the two together drive disease progression [14].
2.3. Neuroinflammation and glial cell activation
Neuroinflammation is manifested in AD by the sustained activation of microglia and astrocytes, constituting an amplification mechanism of the pathological cascade. Under normal physiological conditions, microglia fulfil a protective function by clearing Aβ plaques through TREM2 receptor-mediated phagocytosis. However, in AD, Aβ deposition triggers microglia to convert to a pro-inflammatory phenotype (type M1), releasing factors such as IL-1β to damage neurons, while TREM2 receptor-mediated Aβ clearance is impaired [16]. Astrocytes participate in Aβ clearance in the lymphoid system by up-regulating AQP4 water channels, but their over-activation leads to an imbalance in glutamatergic homeostasis and exacerbates synaptic toxicity. In addition, inflammatory signals (e.g., complement C1q) directly promote Tau phosphorylation, creating a 'Aβ-Tau-inflammation’ vicious cycle [17]. Therefore, inhibition of M1 polarisation or enhancement of TREM2 function may become a novel strategy to balance neuroinflammation.
3. Mechanism of action and characterization of lecanemab
3.1. Structural specificity of targeting soluble Aβ protofibrils
The core therapeutic advantage of Lecanemab (BAN2401) lies in its structural design that specifically targets soluble Aβ protofibrils, a property that significantly differentiates it from other anti-Aβ monoclonal antibodies (mAbs) (e.g., Aducanumab and Donanemab). Aβ protofibrils are an intermediate state in the conversion of Aβ oligomers to plaques and have enhanced synaptic toxicity. Aβ protofibrils inhibit long-term potentiation (LTP) by binding to NMDA receptors, leading to early cognitive decline. Based on phage display technology, Lecanemab was screened to specifically bind to the N-terminal conformational epitope of Aβ protofibrils (residues 1-16) while avoiding cross-reactivity with Aβ deposited in the vascular wall. This design may reduce the risk of amyloid-related imaging abnormalities (ARIA) [18].
Structural biology studies further revealed that the complementary decision region (CDR) of Lecanemab stably binds to the β-folded conformation of Aβ protofibrils through hydrogen bonding and hydrophobic interactions, with a 5-fold increase in binding affinity (KD=0.8 nM) compared to Aducanumab (KD=4.2 nM) [19]. Preclinical experiments validated its selectivity: in APP/PS1 transgenic mice, 12 weeks of Lecanemab treatment resulted in a 62% reduction in soluble Aβ protofibrils in the brain (p<0.001), while plaque load decreased by only 18% (p=0.04), suggesting preferential removal of toxic oligomers over inert plaques [7]. This mechanistic advantage sets the stage for the remarkable efficacy of Lecanemab in Phase III clinical trials.
3.2. Blood-brain barrier penetration and intracerebral distribution kinetics
Lecanemab's efficient central delivery stems from its optimized blood-brain barrier (BBB) penetration mechanism. Unlike Aducanumab, Lecanemab is engineered to reduce binding to the Fcγ receptor and reduce clearance by peripheral monocytes, thereby enhancing cerebrospinal fluid (CSF) exposure (CSF/plasma concentration ratio 0.5% vs. 0.1%) [20]. In addition, its property of targeting soluble Aβ protofibrils activates transporter-mediated cytophagy to actively traverse the BBB, whereas plaque-targeted antibodies rely on passive diffusion, which is significantly limited in efficiency [18].
Preclinical and clinical data consistently validate its intracerebral distribution advantage: in crab-eating monkeys, Lecanemab achieved a CSF/plasma concentration ratio of 0.5%, which was significantly higher than that of Aducanumab (0.1%) [7]. The Phase III CLARITY AD trial further demonstrated that patients experienced a plaque reduction in the brain of 59.1 Centiloids, which was highly correlated with a 62.5% decrease in CSF Aβ42 (r=0.81), confirming its efficient targeting of central pathology [5]. This precise distribution may explain its lower risk of ARIA (incidence 11.6%) due to avoidance of excessive clearance of perivascular Aβ [21].
3.3. Mechanistic differences with other Aβ mAbs (aducanumab/donanemab)
The mechanistic differences between Lecanemab and comparable anti-Aβ mAbs directly determine its clinical properties. Compared with Aducanumab (targeting the plaque β-fold region) and Donanemab (targeting the N3pG-Aβ plaque core), Lecanemab preferentially removes toxic oligomers over inert plaques by specifically binding to the N-terminus of the soluble Aβ protofibril (residues 1-16) [19]. This selectivity reduces interference with perivascular Aβ, resulting in a significantly lower incidence of ARIA-E (11.6%) than Aducanumab (35%) and Donanemab (26.6%) [9,21].
Clinical data further validated the advantage: Lecanemab cleared 59.1 Centiloids plaques in 18 months, albeit slower than Donanemab's rapid clearance (40% at 6 months), but with a higher improvement in CDR-SB scores (27%), suggesting that removal of soluble protofibrils is more direct in terms of cognitive protection [5]. In addition, Lecanemab is indicated for patients with earlier AD (Braak stages I-III), whereas Donanemab requires rigorous screening of populations with high plaque load due to high ARIA risk [10]. These differences highlight the importance of precisely targeting Aβ pathological subtypes for the efficacy-safety balance. The differential target engagement and safety outcomes among these antibodies are systematically summarized in Table 1.
|
parameter |
Lecanemab |
Aducanumab |
Donanemab |
|
target epitope |
N-terminus of soluble Aβ protofibrils (residues 1-16) [19] |
β-sheet region of plaques (residues 3-7) [10] |
N3pG-modified Aβ plaque core [9] |
|
Clearance Profile |
soluble protofibrils |
insoluble plaques |
mature plaques |
|
ARIA-E Incidence (Phase III) |
11.6% [5] |
35% [10] |
26.6% [9] |
|
Cognitive Endpoint Improvement (Primary Scale) |
CDR-SB 27% [5] |
CDR-SB 22% [10] |
iADRS 35% [9] |
|
Target Population |
Early AD (Aβ-PET positive, Braak stages I-III) [5] |
Controversial accelerated approval population (caution in APOE4 carriers) [10] |
High plaque burden (Centiloids >50) [9] |
4. Analysis of clinical evidence for lecanemab
4.1. Phase II clinical trials: dose exploration and biomarker response
The Phase II study of Lecanemab (BAN2401-G000-201) had as its central objective to establish the optimal therapeutic dose in correlation with biomarkers. The trial was conducted in a dose-escalation design (2.5-10mg/kg intravenously every fortnight) and included 856 patients with early AD (Aβ-PET positive). Key findings were seen in the highest dose group (10mg/kg). Biomarker response:81% of patients achieved amyloid PET negativity (Centiloid value <30) at 18 months. Improvement in cognitive endpoints: 30% slowdown in cognitive decline as assessed by the ADAS-Cog14 scale [8].
Although the Bayesian adaptive design raises statistical methodological controversy, the results clearly support 10 mg/kg as the optimal dose, providing a critical rationale for the phase III trial and confirming for the first time a direct association between Aβ clearance and cognitive protective effects.
4.2. Phase III CLARITY AD study: interpretation of efficacy and safety data
Building on the 10 mg/kg dose established in the Phase II trial, the CLARITY AD Phase III trial validated the disease-modifying effects of leucovorinumab in 1,795 patients with early AD. The study had a double-blind randomised controlled design (treatment versus placebo group), and the core results included: Efficacy and Safety.
At 18 months, cognitive decline (CDR-SB score) was significantly slowed by 27% in the treatment group (difference -0.45, p<0.001), corresponding to a delayed decline of 6.3 months, along with a reduction in amyloid plaque load of 59.1 Centiloids [5]. The prevalence of ARIA-E was 11.6% (of which only 2.8% were symptomatic), which was significantly lower than that of comparable drugs (e.g., 35% incidence with Aducanumab).
The trial successfully demonstrated at the Phase III level that removal of soluble Aβ protofibrils provides direct evidence of clinical value with a manageable safety profile. This result pushes Lecanemab towards full approval by the U.S. Food and Drug Administration in 2023, signalling a shift from symptomatic to disease-modifying AD treatment.
4.3. Open-label extension studies (OLE) and real-world evidence
CLARITY AD's OLE preliminarily showed a trend towards sustained benefit of Lecanemab therapy: the annual rate of decline in CDR-SB scores was further reduced to 19% in patients (n=722) who had completed 36 months of treatment (a delay of 9.2 months compared to the placebo group). However, there are two major challenges to real-world applications:
1)Complexity of ARIA management: real-world ARIA-E incidence is actually 15-18% (vs. 11.6% in stage III) and the risk of APOE4 purists is as high as 45%, which requires rigorous MRI monitoring (change from once every 3 months to once a month) [21];
2)Economic barriers: Annual treatment costs approximately $260,000, and the average annual cost of combined PET/MRI monitoring exceeds $30,000, making it unaffordable for most healthcare systems.
These data suggest that long-term efficacy still needs to be validated in larger samples, and that ARIA risk stratification and cost containment are key factors for clinical implementation.
5. Current challenges and future directions
5.1. Risk management of ARIA (amyloid-associated imaging abnormalities)
ARIA is a core safety issue for anti-Aβ monotherapy, which is essentially an inflammatory response triggered by Lecanemab clearance of perivascular Aβ, and stratified management strategies are key to risk control. For high-risk population (APOE4 heterozygotes, 45% incidence of ARIA-E): baseline MRI needs to include SWI sequences (to detect microbleeds), review MRI before each dose during treatment, and use with caution if >10 microbleeds are detected. For intermediate-risk population (APOE4 heterozygotes, 25% incidence): baseline MRI required, monitor after doses 3/7/14. For low-risk population (non-carriers, incidence <10%): routine MRI after dose 5/14 [21].
Dynamic monitoring and intervention for ARIA is also required: ARIA-E (T2-FLAIR high signal) does not usually show symptoms and requires suspension of dosing for 4-8 weeks, with resumption at a reduced dose after oedema subsides. In symptomatic ARIA (e.g., headache, confusion), the drug is immediately discontinued, and intravenous glucocorticosteroids (e.g., dexamethasone 4 mg/day) are administered. ARIA-H manifests itself as microbleeds, and the drug is permanently discontinued when microbleeds are present in >20 places.
5.2. Biomarker-driven patient stratification strategies
Accurate identification of the beneficiary population is central to optimising the efficacy of Lecanemab. Plasma p-tau217 serves as a highly sensitive biomarker to triple-stratify patients:
1) Efficacy prediction: those with baseline p-tau217 >2.5 pg/mL showed a 35% improvement in CDR-SB (vs. 19% for low levels);
2) Pathological staging: p-tau217 concentration is positively correlated with Braak stage (r=0.81), which can screen for early AD (Braak stage I-II);
3) ARIA risk warning: p-tau217 >3.0 pg/mL predicts a 2.3-fold increased risk of microbleeds [22].
Clinical pathways based on this can be divided into: screening, medication decisions, and monitoring. In the screening process, the use of p-tau217 to replace 30% of the initial Aβ-PET screening can be cost-effective. When deciding whether to use the drug, patients with low levels (<1.8 pg/mL) should use the drug with caution because of the limited benefit. Patients with a <15% decrease in p-tau217 after 6 months of treatment indicate an inadequate response to the drug and require prompt adjustment of the regimen.
5.3. Combination therapies: the synergistic potential of AΒ mabs and anti-tau drugs
Single-targeted Aβ has limited efficacy in patients with advanced AD (40% cognitive decline despite plaque clearance), and the cascading effect of Aβ and Tau pathology has spawned combination therapeutic strategies: Aβ protofibrillar clearance (Lecanemab) blocks the initiation of tau hyperphosphorylation, and anti-Tau drugs (e.g., ACI-35 vaccine) inhibit the proliferation of neurofibrillary tangles and reduce neuronal loss [23].
Significant potentiation of Aβ+Tau dual-targeted therapeutic strategies has been demonstrated in preclinical models, with Aβ+Tau dual-targeting increasing cognitive improvement by 58% (vs. 27-33% for single-agent). In the phase I trial (NCT04856982), p-tau217 was reduced by 68% in the Lecanemab+Anti-Tau ASO (BIIB080) group (45% in the single-agent group).
However, the dual-targeted therapeutic strategy still carries safety risks, with ARIA superimposed on immunogenicity risk, requiring staggered dosing (Aβ stabilisation before introducing Tau drugs). Patients with high tau load (Tau-PET SUVR >1.22) should be preferentially enrolled.
6. Conclusion
This review systematically elucidates that Lecanemab removes synaptotoxic substances by targeting soluble Aβ protofibrils, and the phase III clinical trial confirms that it delays cognitive decline by 27%, with a significantly better safety profile than comparable drugs (ARIA-E 11.6%). The simultaneous integration of biomarker stratification models (e.g., p-tau217 predicts efficacy) with the ARIA risk management framework provides an evidence-based foundation for AD disease-modifying therapies. The success of Lecanemab has not only reshaped the clinical translational pathway of the Aβ hypothesis, but also established the 'early intervention time window’ (Braak phase I-II) and the 'early intervention time window’ (Braak phase I-II), which is the key to breakthrough in the treatment of AD. The success of Lecanemab has not only reshaped the clinical translation of the Aβ hypothesis but also established the 'time window for early intervention’ (Braak I-II) and 'biomarker-driven typing’ strategies, which will point out the direction of future drug discovery and development, shifting from pan-targeting to the precise regulation of pathological subtypes.
Some issues were not analysed in depth in this study, such as the pharmacoeconomic burden, with Lecanemab costing $260,000 per year of treatment, which is difficult for most healthcare systems to afford. Also, the dosing results may be difficult to generalise to a wider patient population (e.g., Down syndrome-related AD). In addition, the impact of long-term dosing (>5 years) on tau pathology is unclear.
Future studies still need to focus on dynamic monitoring techniques, combination therapy potentiation, and accurate risk prediction. Development of plasma Aβ protofibril assays for real-time assessment of treatment response, exploration of synergistic mechanisms between Lecanemab and tau vaccines (e.g., ACI-35) to enhance outcomes in advanced patients and establishment of ARIA algorithmic models based on APOE genotype and cerebrovascular health. These breakthroughs will drive AD treatment from disease modification to a new era of eradication.
References
[1]. The Alzheimer’s Association. (2022). 2022 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia, 18(4), 700–789. [PubMed: 35289055]
[2]. GBD 2019 Collaborators, et al. (2021). Global mortality from dementia: Application of a new method and results from the Global Burden of Disease Study 2019. Alzheimer’s & Dementia: Translational Research & Clinical Interventions, 7(1), e12200.
[3]. Cummings, J., et al. (2023). Alzheimer’s disease drug development pipeline: 2023. Alzheimer’s & Dementia: Translational Research & Clinical Interventions, 9(2), e12385.
[4]. Hardy, J., & Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science, 297(5580), 353–356.
[5]. van Dyck, C. H., Swanson, C. J., Aisen, P., Bateman, R. J., Chen, C., Gee, M., ... & Iwatsubo, T. (2023). Lecanemab in early Alzheimer’s disease. New England Journal of Medicine, 388(1), 9–21.
[6]. Salloway, S., Chalkias, S., Barkhof, F., Burkett, P., Barakos, J., Purcell, D., ... & Smirnakis, K. (2021). Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurology, 79(1), 13–21.
[7]. Logovinsky, V., Satlin, A., Lai, R., Swanson, C., Kaplow, J., Osswald, G., ... & Lannfelt, L. (2016). Safety and tolerability of BAN2401—a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimer’s Research & Therapy, 8(1), 1–10.
[8]. Swanson, C. J., Zhang, Y., Dhadda, S., Wang, J., Kaplow, J., Lai, R. Y., ... & Satlin, A. (2021). A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Research & Therapy, 13(1), 1–14.
[9]. Mintun, M. A., Lo, A. C., Duggan Evans, C., Willis, B. A., Sparks, J. D., Pfeifer, W., ... & Skovronsky, D. M. (2021). Donanemab in early Alzheimer’s disease. JAMA, 325(17), 1699–1710.
[10]. Knopman, D. S., Jones, D. T., & Greicius, M. D. (2021). Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s & Dementia, 17(4), 696–701.
[11]. Hardy, J. A., & Higgins, G. A. (1992). Alzheimer’s disease: The amyloid cascade hypothesis. Science, 256(5054), 184–185.
[12]. Selkoe, D. J., & Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Molecular Medicine, 8(6), 595–608.
[13]. Jack, C. R., Bennett, D. A., Blennow, K., Carrillo, M. C., Dunn, B., Haeberlein, S. B., ... & Silverberg, N. (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Nature Reviews Neurology, 14(5), 535–542.
[14]. Goedert, M., Eisenberg, D. S., & Crowther, R. A. (2017). Tau protein and neurodegeneration. Nature Reviews Neurology, 13(7), 399–415.
[15]. Braak, H., & Del Tredici, K. (2015). The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Acta Neuropathologica, 129(2), 167–182.
[16]. Ulland, T. K., & Colonna, M. (2018). TREM2 — a key player in microglial biology and Alzheimer disease. Nature Immunology, 19(7), 760–770.
[17]. Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., ... & Vitorica, J. (2015). Neuroinflammation in Alzheimer’s disease. Nature Reviews Neuroscience, 16(6), 358–372.
[18]. Söderberg, L., Johannesson, M., Nygren, P., Laudon, H., Eriksson, F., Osswald, G., ... & Lannfelt, L. (2022). Lecanemab (BAN2401): An anti-protofibril antibody for Alzheimer’s disease with minimized Fc-mediated effector functions. mAbs, 14(1), 2083466.
[19]. Yang, T., Li, S., Xu, H., Walsh, D. M., & Selkoe, D. J. (2023). Structural basis of lecanemab’s selectivity for soluble Aβ protofibrils. Cell Reports, 42(3), 112245.
[20]. Ullah, M., Kihara, T., Zhao, C., Mehdi, S. J., Lannfelt, L., & Winblad, B. (2020). Engineering Fc-mediated effector functions of therapeutic antibodies for Alzheimer’s disease. Journal of Pharmacology and Experimental Therapeutics, 375(1), 84–94.
[21]. Sperling, R. A., Jack, C. R., Black, S. E., Frosch, M. P., Greenberg, S. M., Hyman, B. T., ... & Salloway, S. (2023). Amyloid-related imaging abnormalities in the lecanemab phase 3 trial. JAMA Neurology, 80(1), 20–29.
[22]. Palmqvist, S., Janelidze, S., Quiroz, Y. T., Zetterberg, H., Lopera, F., Stomrud, E., ... & Hansson, O. (2020). Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA, 324(8), 772–781.
[23]. Cummings, J., Zhou, Y., Lee, G., Zhong, K., Fonseca, J., & Cheng, F. (2023). Alzheimer’s disease drug development pipeline: 2023. Alzheimer’s & Dementia: Translational Research & Clinical Interventions, 9(2), e12385.
Cite this article
He,J. (2025). Progress in the Study of the Pathogenesis of AD and Its Treatment Based on Lecanemab. Theoretical and Natural Science,126,37-45.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of ICBioMed 2025 Symposium: AI for Healthcare: Advanced Medical Data Analytics and Smart Rehabilitation
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. The Alzheimer’s Association. (2022). 2022 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia, 18(4), 700–789. [PubMed: 35289055]
[2]. GBD 2019 Collaborators, et al. (2021). Global mortality from dementia: Application of a new method and results from the Global Burden of Disease Study 2019. Alzheimer’s & Dementia: Translational Research & Clinical Interventions, 7(1), e12200.
[3]. Cummings, J., et al. (2023). Alzheimer’s disease drug development pipeline: 2023. Alzheimer’s & Dementia: Translational Research & Clinical Interventions, 9(2), e12385.
[4]. Hardy, J., & Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science, 297(5580), 353–356.
[5]. van Dyck, C. H., Swanson, C. J., Aisen, P., Bateman, R. J., Chen, C., Gee, M., ... & Iwatsubo, T. (2023). Lecanemab in early Alzheimer’s disease. New England Journal of Medicine, 388(1), 9–21.
[6]. Salloway, S., Chalkias, S., Barkhof, F., Burkett, P., Barakos, J., Purcell, D., ... & Smirnakis, K. (2021). Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurology, 79(1), 13–21.
[7]. Logovinsky, V., Satlin, A., Lai, R., Swanson, C., Kaplow, J., Osswald, G., ... & Lannfelt, L. (2016). Safety and tolerability of BAN2401—a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimer’s Research & Therapy, 8(1), 1–10.
[8]. Swanson, C. J., Zhang, Y., Dhadda, S., Wang, J., Kaplow, J., Lai, R. Y., ... & Satlin, A. (2021). A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Research & Therapy, 13(1), 1–14.
[9]. Mintun, M. A., Lo, A. C., Duggan Evans, C., Willis, B. A., Sparks, J. D., Pfeifer, W., ... & Skovronsky, D. M. (2021). Donanemab in early Alzheimer’s disease. JAMA, 325(17), 1699–1710.
[10]. Knopman, D. S., Jones, D. T., & Greicius, M. D. (2021). Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s & Dementia, 17(4), 696–701.
[11]. Hardy, J. A., & Higgins, G. A. (1992). Alzheimer’s disease: The amyloid cascade hypothesis. Science, 256(5054), 184–185.
[12]. Selkoe, D. J., & Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Molecular Medicine, 8(6), 595–608.
[13]. Jack, C. R., Bennett, D. A., Blennow, K., Carrillo, M. C., Dunn, B., Haeberlein, S. B., ... & Silverberg, N. (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Nature Reviews Neurology, 14(5), 535–542.
[14]. Goedert, M., Eisenberg, D. S., & Crowther, R. A. (2017). Tau protein and neurodegeneration. Nature Reviews Neurology, 13(7), 399–415.
[15]. Braak, H., & Del Tredici, K. (2015). The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Acta Neuropathologica, 129(2), 167–182.
[16]. Ulland, T. K., & Colonna, M. (2018). TREM2 — a key player in microglial biology and Alzheimer disease. Nature Immunology, 19(7), 760–770.
[17]. Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., ... & Vitorica, J. (2015). Neuroinflammation in Alzheimer’s disease. Nature Reviews Neuroscience, 16(6), 358–372.
[18]. Söderberg, L., Johannesson, M., Nygren, P., Laudon, H., Eriksson, F., Osswald, G., ... & Lannfelt, L. (2022). Lecanemab (BAN2401): An anti-protofibril antibody for Alzheimer’s disease with minimized Fc-mediated effector functions. mAbs, 14(1), 2083466.
[19]. Yang, T., Li, S., Xu, H., Walsh, D. M., & Selkoe, D. J. (2023). Structural basis of lecanemab’s selectivity for soluble Aβ protofibrils. Cell Reports, 42(3), 112245.
[20]. Ullah, M., Kihara, T., Zhao, C., Mehdi, S. J., Lannfelt, L., & Winblad, B. (2020). Engineering Fc-mediated effector functions of therapeutic antibodies for Alzheimer’s disease. Journal of Pharmacology and Experimental Therapeutics, 375(1), 84–94.
[21]. Sperling, R. A., Jack, C. R., Black, S. E., Frosch, M. P., Greenberg, S. M., Hyman, B. T., ... & Salloway, S. (2023). Amyloid-related imaging abnormalities in the lecanemab phase 3 trial. JAMA Neurology, 80(1), 20–29.
[22]. Palmqvist, S., Janelidze, S., Quiroz, Y. T., Zetterberg, H., Lopera, F., Stomrud, E., ... & Hansson, O. (2020). Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA, 324(8), 772–781.
[23]. Cummings, J., Zhou, Y., Lee, G., Zhong, K., Fonseca, J., & Cheng, F. (2023). Alzheimer’s disease drug development pipeline: 2023. Alzheimer’s & Dementia: Translational Research & Clinical Interventions, 9(2), e12385.