







1. Introduction
Glutathione (GSH) is an important tripeptide in cellular signaling and antioxidant defenses [1]. In previous studies, the primary role of GSH is to as an antioxidant scavenging Reactive Oxygen Species (ROS) and modulating Nitric Oxide Synthase (NOS) activity, which can reduce damage to cell caused by ROS and NOS. For people with ASD, the level of GSH is much lower than average. This results in more frequent mitochondria damage in, leading to inefficiency in impulse transmission. Also, due to deficiency in GSH synthesis, glutamate (Glu), substrate in GSH synthesis, accumulates and causes neuroinflammation. Current treatments primarily focus on GSH level elevation or substitutions of GSH to perform analogous functions. These treatments are promising in curing ASD and other neurodegenerative disorders with similar pathogenesis. This paper provides a systematic review of different treatments involving their mechanism and clinical data, which is significant in comparing effects of different treatments and giving a more narrow-down treating mindset.
2. Pathogenesis
GCL is a primary enzyme in GSH synthesis used to catalyst bond between glutamate and cysteine, which is the rate limiting step in GSH synthesis [1,2]. As observed in ASD children, both cysteine and GCL level is lower than normal, leading to inhibition of GCL activity, which is a main cause of reducing GSH level. Due to the inhibition of GCL activity, glutamate, the substrate of rate limiting step will accumulate, which will further inhibit GSH synthesis and cause neurotoxicity.
GSH/oxidised glutathione (GSSG) ratio in ASD patients is also lower than normal. Previous research shows that GSH/GSSG ratio and content of GSH in the lymphoblastoid cells, brain, and blood tissues of ASD patients are lower than average[2]. This imbalance will result in oxidative stress and cellular dysfunction.
3. Treatment based on glutathione deficiency
3.1. Increase GCL level to accelerate GSH synthesis
3.1.1. Mechanism of bypassing GCL treatment
Gamma-glutamylcysteine, an intermediate in GSH synthesis, emerges as a promising therapeutic strategy to elevate intracellular GSH levels by bypassing GCL[3]. GCL catalyst the formation of γ-glutamyl bond by condensation of γ-COOH and α-NH2 in cysteine and glutamate to produce γ-GC, which is the product of rate determine step in GSH synthesis. Delivering of γ-GC to ASD patients can bypass the rate limiting step, directly providing sufficient γ-GC for the subsequent steps. γ-GC is actively or passively transported into cells via dipeptide transporters and is efficiently converted to GSH as long as glycine and ATP are available.
3.1.2. Clinical trial bypassing GCL
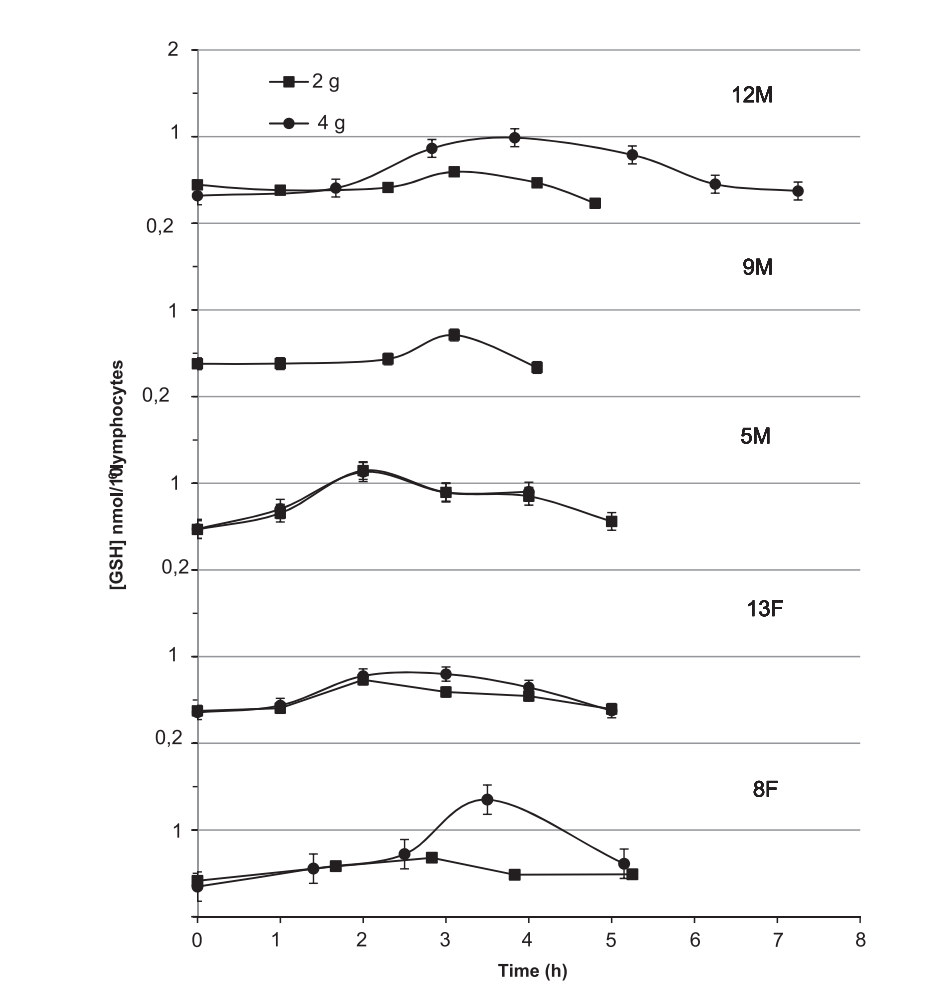
Martin Hani Zarka, Wallace John Bridge’s research [4] shows that oral GGS give rise to GSH level in lymphocytes, but the effects do not last long. The experiment involving double-blind crossover trial (n=6) and pre-post study (n=13) tested single oral doses (2 g and 4 g) of GGS or placebo in healthy adults (25–70 years) under non-fasting conditions to avoid substrate limitation. As it is shown in Figure 1, blood samples assessed lymphocyte GSH levels before dose and up to 7 hours after treatment. In all five groups, the level of GSH increases from 2-4 hours, after that followed by a decline to the original level from 4-7 hour. The maximum increase of GSH is higher in 4g group. The treatment is effective but the duration of GSH elevation still needs to be improved. Also, the experiment testing concentration of GSH in lymphocytes which locate in all parts of body except brain. Whether oral GGC can elevate GSH concentration in brain (e.g. glial) needs to be discussed.
3.1.3. Mechanism of insulin-like growth factor 1 in regulating GCL synthesis
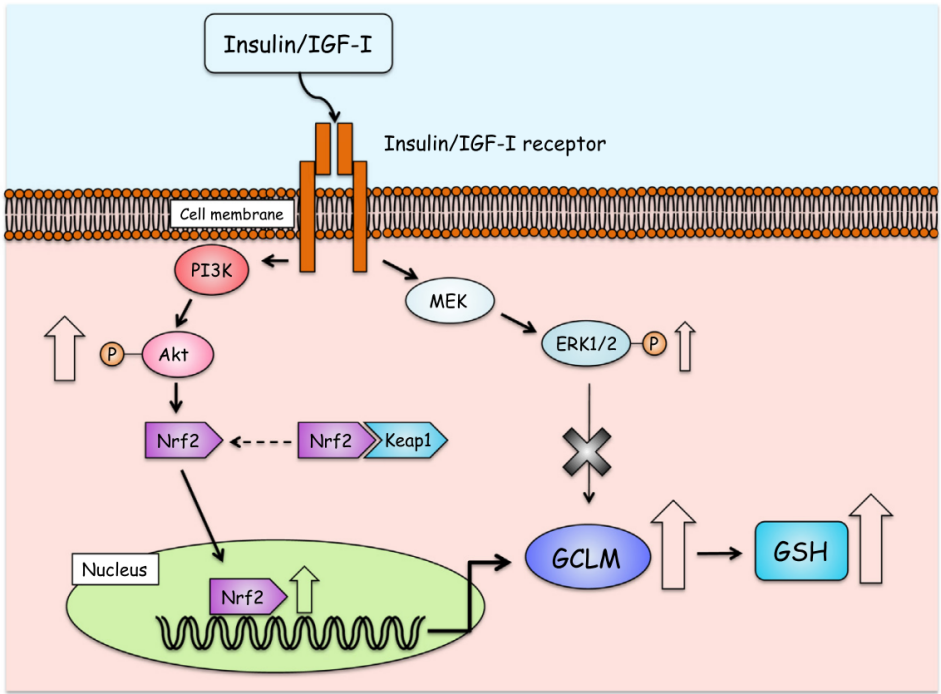
As glutamate-cysteine ligase is the rate-limiting enzyme in production of GSH, increasing activity of GCL can accelerate synthesis of GSH. The transcriptional regulation of glutamate-cysteine ligase involves multiple transcription factors binding to promoter elements, and c-Myc[3]. AP-1 and ARE are central to oxidative stress-induced upregulation [5]. Nrf2 is the primary regulator, binding AREs to activate antioxidant genes under oxidative stress. Under basal conditions, Nrf2 is sequestered by Keap1 for degradation, but oxidative modifications trigger Nrf2 nuclear translocation. Nrf1 supports basal Gclm expression, while c-Myc and TNFα-mediated pathways further modulate subunit expression. This coordinated regulation ensures GCL-mediated GSH synthesis adapts to redox challenges. As shown in Figure 2[6], insulin-like growth factor 1 (IGF-1) acting as a transcriptional factor, binding to IGF-1 receptor on cells membrane of SH-SY5Y , and activates phosphoinositide 3-kinase (PI3K), which induces phosphorylation of protein kinase B (Akt) , which regulates activity of downstream effectors including Nrf2, resulting in increased mRNA expression for GCLM in SH-SY5Y cells.
3.1.4. Clinical trial of insulin-like growth factor 1 treatment
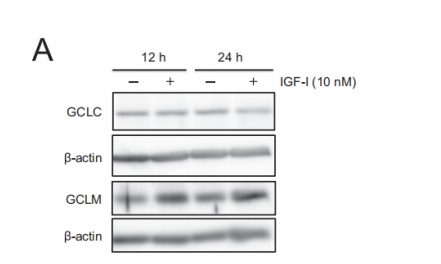
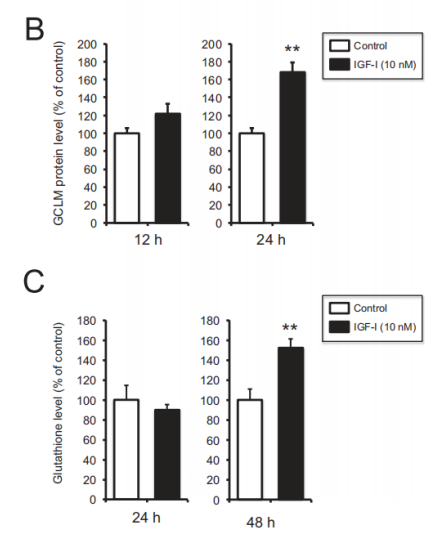
In Shuhei Takahashi, Akinori Hisatsune et al. [7] found that IGF-1 can elevate level of GSH in SH-SY5Y cells. In the study, SH-SY5Y cells were divided into vehicle control or IGF-1 treatment groups. A fraction of scraped cells was used for cell counting. Subsequently, the absorbance at 450 nm was measured. As is shown in Figure 3, Gamma-glutamyl cysteine ligase modifier subunit (GCLM) increase when treated with IGF-1 for both 12 hours and 24 hours, while no change in content of Gamma-glutamyl cysteine ligase catalytic subunit (GCLC). The increase of GCLM concentration is more obvious in 24-hour treatment. For glutathione level, no significant change in 24-hour treatment due to the overlap of error bar in graph C, and the elevation is significant when treatment duration increases to 48 hours.
3.2. Re-balance glutathione/glutamate ratio to avoid inhibition of GSH synthesis
3.2.1. Mechanism of n-acetylcysteine in re-balancing gsh/glu
NAC (N-acetylcysteine) is the acetylated form of L-cysteine, which is rapidly absorbed orally. Once absorbed, as shown in Figure 4, L-cysteine is oxidized to cystine in the brain’s prooxidant environment[8]. Cystine serves as a substrate for the cystine-glutamate antiporter (xCT system), a transporter primarily expressed in astrocytes. This antiporter exchanges intracellular cystine for extracellular glutamate, enabling cysteine uptake into cells. Inside the cell, cystine is reduced back to cysteine, a critical component for synthesizing GSH (GSH), the body’s primary antioxidant. NAC’s dual role—modulating the cystine-glutamate antiporter to control glutamate homeostasis and boosting GSH production—underlies its therapeutic effects. By reducing excess glutamate (preventing NMDA receptor overactivation) and combating oxidative stress via GSH, NAC shows promise in treating neuropsychiatric disorders.
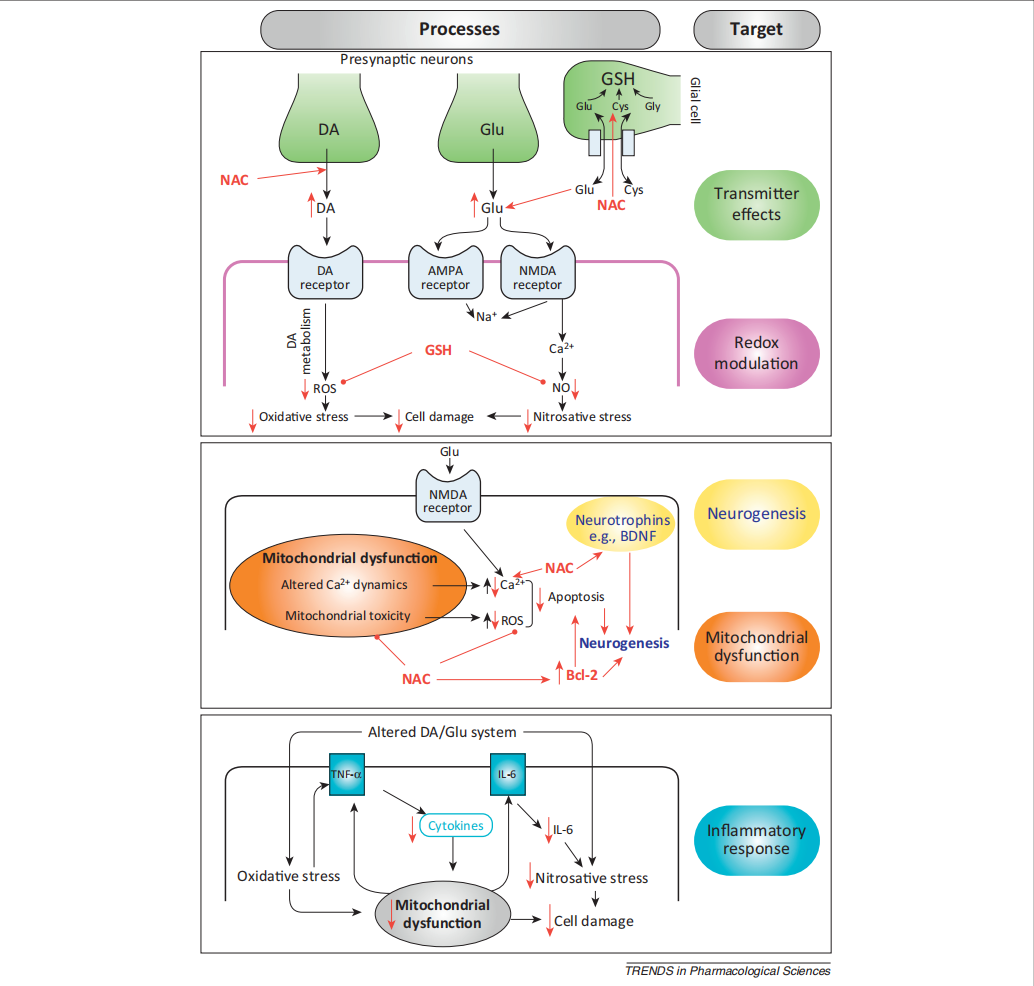
3.2.2. Clinical trial of n-acetylcysteine treatment
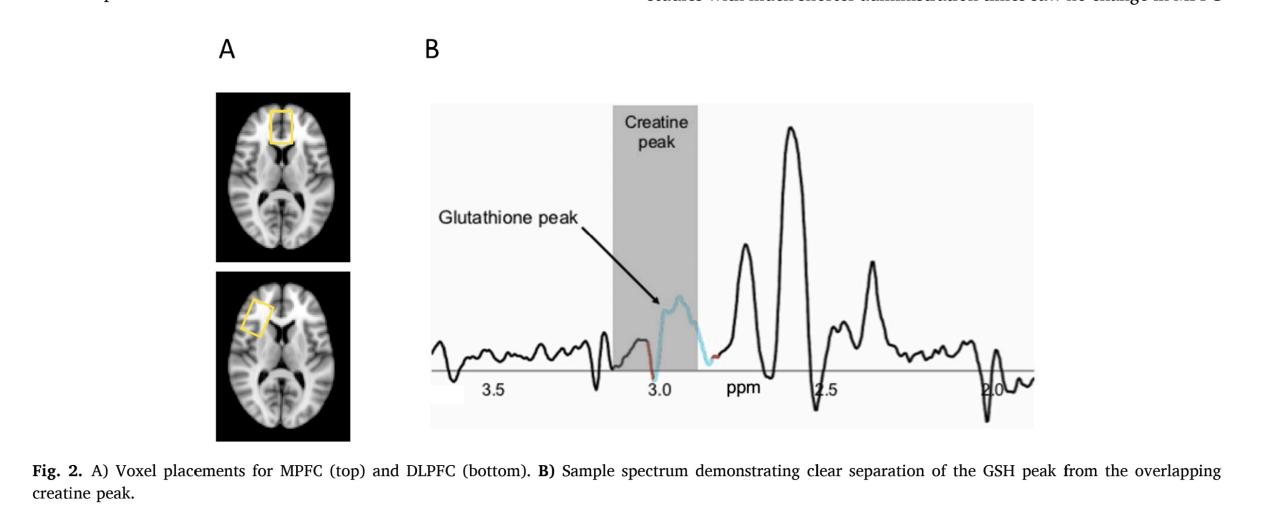
In trial experiment of Junghee Lee et al. 40 patients (21 NAC, 19 placebo) received oral NAC (2400 mg/day: 1200 mg twice daily) or placebo for 8 weeks [9]. For measurement of dependent variable, use LC-MS to measure concentration of GSH in blood plasma at baseline and 8 weeks, the result of LC-MS will be analysis by isotope-labeled. Researchers used magnetic resonance spectroscopy (MRS) to measure glutamate and GSH levels in medial and dorsolateral prefrontal cortex (MPFC, DLPFC) in Figure 5 using blood plasma sample from Peripheral, the content is normalised to creatine ratio.
As it is shown in Figure 6 and Figure 7, in MPFC, elevation in GSH level is significant before and after taking NAC compared to placebo, (p=0.035, which is less than 0.05), and there is a trend level decrease in glutamate level (p=0.054, which is more than 0.05 but still less than 0.1). In DLPFC, no significant increase in GSH level (p=0.88) and no significant decrease in glutamate level (p=0.134), which means that the effect of NAC treatment differs from region of brain. Whether it can be used to treat ASD still needs to be discussed as the location of GSH/GSSG imbalance in brain is important.
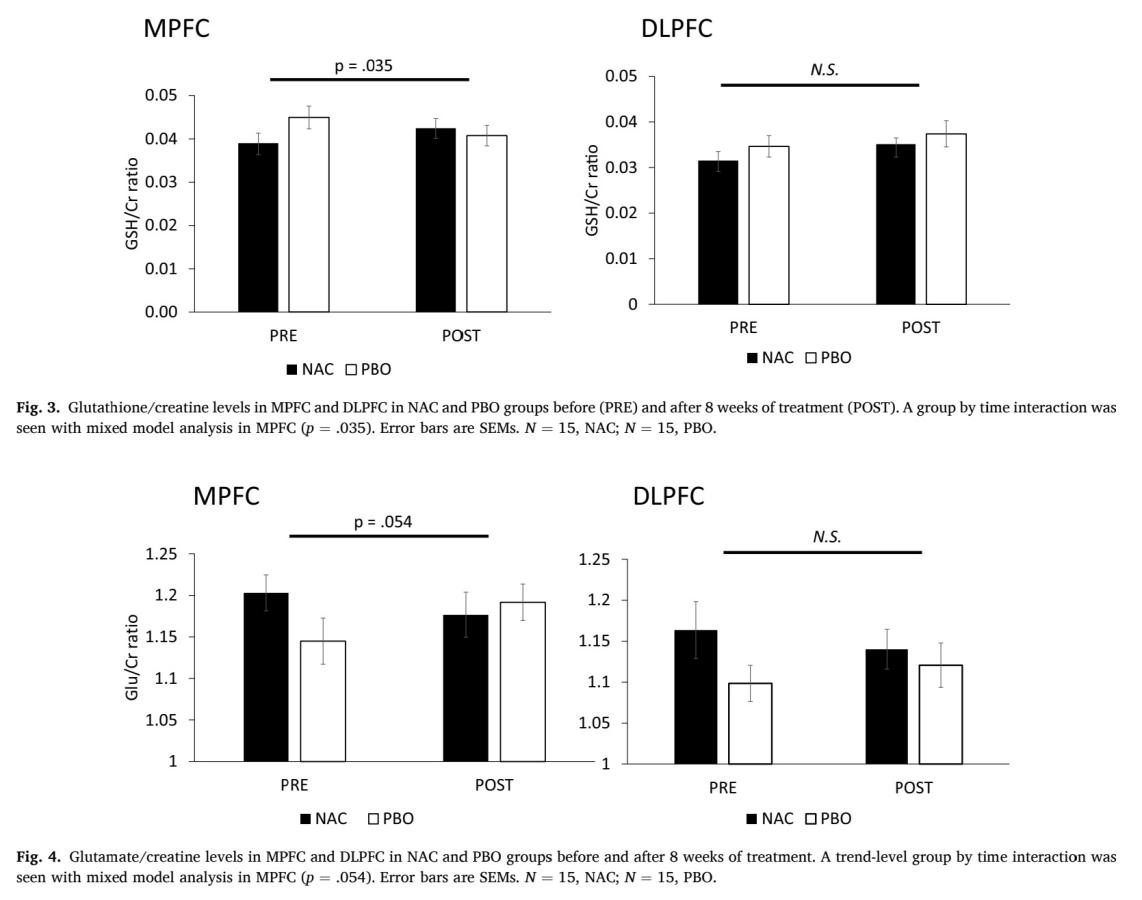
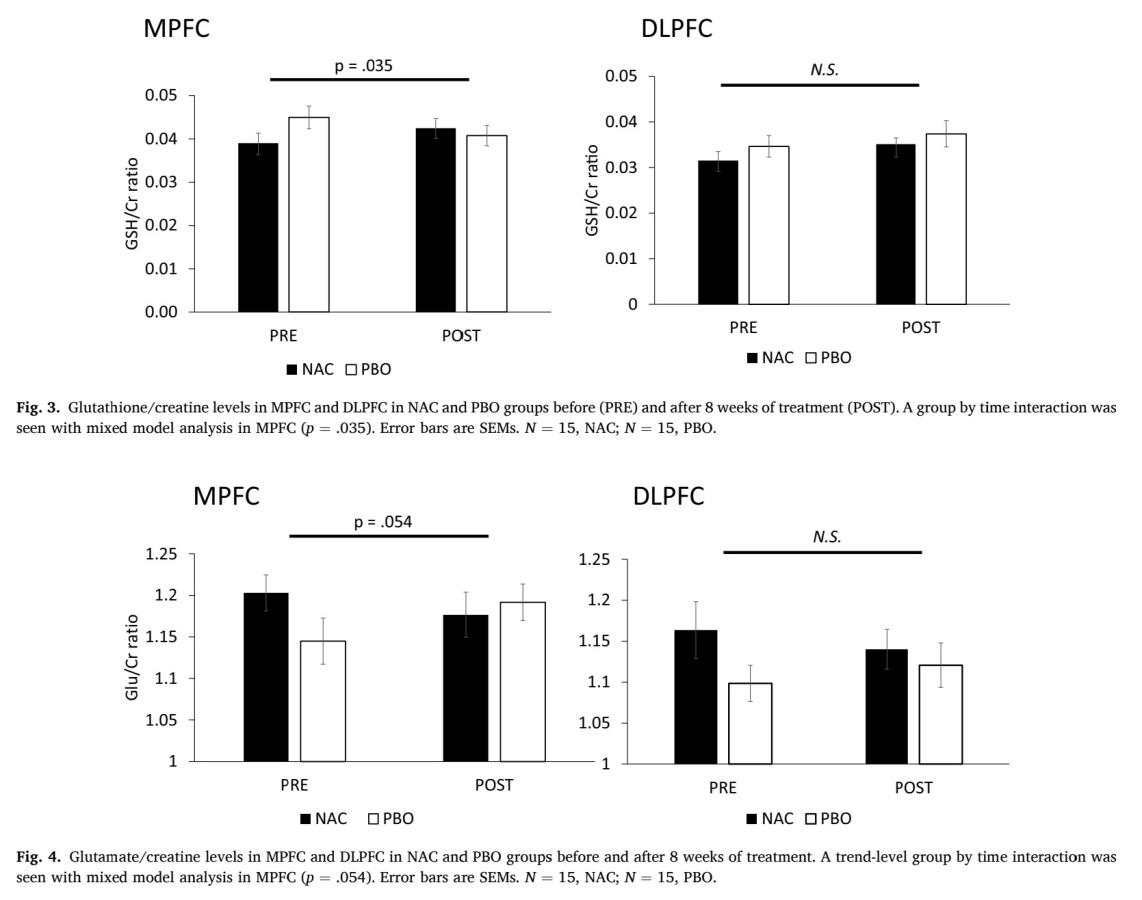
4. Discussion
Though many treatments based on elevation of GSH level are promising in relieving ASD symptoms, there are still to be discussed. According to rodent models, γ-GC treatment gives rise to GSH level in kidney and specific part of brain stem, but the effects on cells that are specific in brain remains unknown. Also, oral γ-GC can be absorbed in intestine, the overall bioavailability and metabolism still needed to be studied systematically. According to the study of Shuhei Takahashi et al. (2015) [7], though IGF-1 can increase expression of GCL and activate glutathione biosynthetic pathway in neuroblastoma SH-SY5Y cells, cellular mechanisms of IGF-1 is different in other cell types. In IGF-1, IGF-1 increase the expression of GCLM but no of GCLC, which is totally contract in hepatocytes [10]. The effect of NAC treatment differs from region of brain. Whether it can be used to treat ASD still needs to be discussed as treatment may be not effective for all region of brain. Clinical study did not find any main effects of group, any treatment effects in the DLPFC, or any effects on GSH peripherally, though the treatment is effective in MPFC. Though NAC can increase level of GSH in specific part of brain, no relieving in symptoms of any neuron disorder is detected as it is indicated in the study that no correlations between brain levels of GSH or glutamate with negative symptoms or cognitive performance, following correction for multiple comparisons. The dose of NAC drug is a crucial factor in determining the effect of therapy. Two studies with much shorter treating times than the previous research shows no increase in even MPFC GSH levels. In Coles et al. (2018), four weeks of high dose (6000 mg/day) NAC gave no change to brain GSH measured with MRS and, in Girgis et al., 2019, an acute oral NAC challenge of 2400 mg did not produce a detectable increase in GSH in 19 patients with schizophrenia. Also, most of these treatments haven’t been clinically tested on large sample of ASD patients, only in small sample or other neuron disorder with similar pathogenesis involving lack of GSH.
5. Conclusion
In conclusion, decrease in GCL level and cysteine level results in insufficient synthesis of GSH, accumulation of another substrate of rate limiting step, glutamate further inhibits GSH synthesis. Also, decrease in GSH/GSSG ratio will cause imbalance in redox reactions. Current treatments based on GCL including GGS and IGF-1 both have been tested clinically, the increase of GSH level caused by GGS ceased rapidly, and the IGF-1 treatment need relatively long exposure time for cells in IGF-1 to give an obvious increase in GSH concentration. Treatment based on GSH/glutamate ratio including NAC, which gives different effects on elevation of GSH level highly depends on position of the cells. Clinical trial data shows that all these treatments have potential in curing ASD, but all of them needed to be improved and their effects on ASD symptoms instead of biochemical substrate data should be tested. Also, therapeutic efficacy requires quantitative evaluation to determine whether such interventions can alleviate autism spectrum disorder (ASD) symptoms in large sample size. The target cell types and therapeutic efficacy across different cell populations for these therapies must be fully defined and quantified. Also, as the specificity to cells is various for different therapies, the combination of different drugs can be a new therapeutic idea. Drug-drug interactions, Optimal dosing ratios, Co-delivery mechanisms are key points that should be determined in further research. These therapeutic not only have potential in releasing ASD symptoms, they also can be effective in relieving other neuron disorders with similar pathogenesis to ASD like Alzheimer's disease, bipolar disorder, which are all clinically observed involving lack of GSH and having GSH/GSSG imbalance.
References
[1]. Geir Bjørklund, et al. “The Role of Glutathione Redox Imbalance in Autism Spectrum Disorder: A Review.” Free Radical Biology and Medicine, vol. 160, 20 Nov. 2020, pp. 149–162, www.sciencedirect.com/science/article/abs/pii/S0891584920311539, https: //doi.org/10.1016/j.freeradbiomed.2020.07.017. Accessed 26 Jan. 2022.
[2]. Geir Bjørklund, et al. “The Impact of Glutathione Metabolism in Autism Spectrum Disorder.” Pharmacological Research, vol. 166, Apr. 2021, p. 105437, https: //doi.org/10.1016/j.phrs.2021.105437. Accessed 11 June 2021.
[3]. Ferguson, Gavin, and Wallace Bridge. “Glutamate Cysteine Ligase and the Age-Related Decline in Cellular Glutathione: The Therapeutic Potential of γ-Glutamylcysteine.” Archives of Biochemistry and Biophysics, vol. 593, Mar. 2016, pp. 12–23, https: //doi.org/10.1016/j.abb.2016.01.017.
[4]. Zarka, Martin, and Wallace Bridge. “Oral Administration of γ-Glutamylcysteine Increases Intracellular Glutathione Levels above Homeostasis in a Randomised Human Trial Pilot Study.” Redox Biology, vol. 11, 1 Apr. 2017, pp. 631–636, www.sciencedirect.com/science/article/pii/S2213231716303718, https: //doi.org/10.1016/j.redox.2017.01.014. Accessed 16 Sept. 2021.
[5]. Bassil, Fares, et al. “Insulin, IGF-1 and GLP-1 Signaling in Neurodegenerative Disorders: Targets for Disease Modification?” Progress in Neurobiology, vol. 118, 1 July 2014, pp. 1–18, www.sciencedirect.com/science/article/pii/S030100821400029X?via%3Dihub, https: //doi.org/10.1016/j.pneurobio.2014.02.005.
[6]. Bailey-Downs, Lora C, et al. Liver-Specific Knockdown of IGF-1 Decreases Vascular Oxidative Stress Resistance by Impairing the Nrf2-Dependent Antioxidant Response: A Novel Model of Vascular Aging. Vol. 67A, no. 4, 1 Apr. 2012, pp. 313–329, https: //doi.org/10.1093/gerona/glr164. Accessed 4 June 2023.
[7]. Takahashi, Shuhei, et al. “Insulin-like Growth Factor 1 Specifically Up-Regulates Expression of Modifier Subunit of Glutamate-Cysteine Ligase and Enhances Glutathione Synthesis in SH-SY5Y Cells.” European Journal of Pharmacology, vol. 771, 11 Dec. 2015, pp. 99–106, www.sciencedirect.com/science/article/pii/S0014299915304064, https: //doi.org/10.1016/j.ejphar.2015.12.013.
[8]. Berk, Michael, et al. “The Promise of N-Acetylcysteine in Neuropsychiatry.” Trends in Pharmacological Sciences, vol. 34, no. 3, Mar. 2013, pp. 167–177, www.cell.com/trends/pharmacological-sciences/fulltext/S0165-6147(13)00002-3?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0165614713000023%3Fshowall%3Dtrue , https: //doi.org/10.1016/j.tips.2013.01.001. Accessed 30 Apr. 2019.
[9]. Yang, Yvonne S, et al. “N-Acetylcysteine Effects on Glutathione and Glutamate in Schizophrenia: A Preliminary MRS Study.” Psychiatry Research: Neuroimaging, vol. 325, 1 Sept. 2022, pp. 111515–111515, https: //doi.org/10.1016/j.pscychresns.2022.111515. Accessed 12 Feb. 2024.
[10]. CAI, Jiaxin, et al. “Differential Regulation of γ-Glutamylcysteine Synthetase Heavy and Light Subunit Gene Expression.” Biochemical Journal, vol. 326, no. 1, 15 Aug. 1997, pp. 167–172, https: //doi.org/10.1042/bj3260167.
Cite this article
Wang,H. (2025). Treatments for Autism Spectrum Disorder Based on Glutathione Level Regulation. Theoretical and Natural Science,126,174-182.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of ICBioMed 2025 Symposium: AI for Healthcare: Advanced Medical Data Analytics and Smart Rehabilitation
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Geir Bjørklund, et al. “The Role of Glutathione Redox Imbalance in Autism Spectrum Disorder: A Review.” Free Radical Biology and Medicine, vol. 160, 20 Nov. 2020, pp. 149–162, www.sciencedirect.com/science/article/abs/pii/S0891584920311539, https: //doi.org/10.1016/j.freeradbiomed.2020.07.017. Accessed 26 Jan. 2022.
[2]. Geir Bjørklund, et al. “The Impact of Glutathione Metabolism in Autism Spectrum Disorder.” Pharmacological Research, vol. 166, Apr. 2021, p. 105437, https: //doi.org/10.1016/j.phrs.2021.105437. Accessed 11 June 2021.
[3]. Ferguson, Gavin, and Wallace Bridge. “Glutamate Cysteine Ligase and the Age-Related Decline in Cellular Glutathione: The Therapeutic Potential of γ-Glutamylcysteine.” Archives of Biochemistry and Biophysics, vol. 593, Mar. 2016, pp. 12–23, https: //doi.org/10.1016/j.abb.2016.01.017.
[4]. Zarka, Martin, and Wallace Bridge. “Oral Administration of γ-Glutamylcysteine Increases Intracellular Glutathione Levels above Homeostasis in a Randomised Human Trial Pilot Study.” Redox Biology, vol. 11, 1 Apr. 2017, pp. 631–636, www.sciencedirect.com/science/article/pii/S2213231716303718, https: //doi.org/10.1016/j.redox.2017.01.014. Accessed 16 Sept. 2021.
[5]. Bassil, Fares, et al. “Insulin, IGF-1 and GLP-1 Signaling in Neurodegenerative Disorders: Targets for Disease Modification?” Progress in Neurobiology, vol. 118, 1 July 2014, pp. 1–18, www.sciencedirect.com/science/article/pii/S030100821400029X?via%3Dihub, https: //doi.org/10.1016/j.pneurobio.2014.02.005.
[6]. Bailey-Downs, Lora C, et al. Liver-Specific Knockdown of IGF-1 Decreases Vascular Oxidative Stress Resistance by Impairing the Nrf2-Dependent Antioxidant Response: A Novel Model of Vascular Aging. Vol. 67A, no. 4, 1 Apr. 2012, pp. 313–329, https: //doi.org/10.1093/gerona/glr164. Accessed 4 June 2023.
[7]. Takahashi, Shuhei, et al. “Insulin-like Growth Factor 1 Specifically Up-Regulates Expression of Modifier Subunit of Glutamate-Cysteine Ligase and Enhances Glutathione Synthesis in SH-SY5Y Cells.” European Journal of Pharmacology, vol. 771, 11 Dec. 2015, pp. 99–106, www.sciencedirect.com/science/article/pii/S0014299915304064, https: //doi.org/10.1016/j.ejphar.2015.12.013.
[8]. Berk, Michael, et al. “The Promise of N-Acetylcysteine in Neuropsychiatry.” Trends in Pharmacological Sciences, vol. 34, no. 3, Mar. 2013, pp. 167–177, www.cell.com/trends/pharmacological-sciences/fulltext/S0165-6147(13)00002-3?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0165614713000023%3Fshowall%3Dtrue , https: //doi.org/10.1016/j.tips.2013.01.001. Accessed 30 Apr. 2019.
[9]. Yang, Yvonne S, et al. “N-Acetylcysteine Effects on Glutathione and Glutamate in Schizophrenia: A Preliminary MRS Study.” Psychiatry Research: Neuroimaging, vol. 325, 1 Sept. 2022, pp. 111515–111515, https: //doi.org/10.1016/j.pscychresns.2022.111515. Accessed 12 Feb. 2024.
[10]. CAI, Jiaxin, et al. “Differential Regulation of γ-Glutamylcysteine Synthetase Heavy and Light Subunit Gene Expression.” Biochemical Journal, vol. 326, no. 1, 15 Aug. 1997, pp. 167–172, https: //doi.org/10.1042/bj3260167.