







1. Introduction
The Michaelis constant (Km) refers to the substrate concentration at which an enzyme-catalyzed reaction proceeds at half of its maximum rate. It reflects the affinity between an enzyme and its substrate and has significant implications for catalytic efficiency and substrate specificity. Accurately determining the Km value of an enzyme is essential for understanding its biochemical properties and for optimizing bioprocesses. However, experimental measurement of enzyme Km values is both challenging and time-consuming. As a result, current biological databases contain only a limited number of experimentally determined Km values. Therefore, developing a fast and accurate method for predicting enzyme Km values is of great importance.
With the advancement of computational technology, researchers have begun to explore the use of computer-based methods to predict enzymatic kinetic parameters. Borger et al [1]. demonstrated that statistical approaches can be applied to predict enzyme kinetic properties. Heckmann et al [2]. successfully used machine learning techniques to predict the catalytic constants of enzymes in Escherichia coli. More recently, Kroll et al [3]. combined deep learning and machine learning methods to achieve efficient prediction of enzyme Km values. They represented protein sequences using UniRep vectors [4], integrated these with molecular fingerprints of substrates, and trained an Extreme Gradient Boosting (XGBoost) model [5], achieving significant improvements in prediction accuracy. This work has provided valuable insights and guidance for subsequent studies. Han et al [6]. developed UniKP, a framework for predicting enzymatic kinetic parameters based on pretrained language models. UniKP effectively integrates the protein language model ProT5-XL-UniRef50 [7] with the substrate language model SMILES Transformer [8], leveraging the powerful capabilities of deep learning to achieve precise predictions. The predictive performance of UniKP on Km values was comparable to that of Kroll et al., demonstrating high accuracy.
Although the above methods have achieved excellent performance in predicting enzyme kinetic parameters, they generally rely on inferring three-dimensional structural information from protein sequences. However, directly using the enzyme’s structural data to extract its 3D information is not only more straightforward but also potentially more accurate. This is because enzyme-substrate interactions primarily occur within the catalytic domain—this domain is not only a critical structural component of enzymes and determinant of substrate specificity, but also has a direct impact on Km values. By focusing on the features of the catalytic domain when predicting Km values, it is possible to reduce the “noise” introduced by non-essential regions of the enzyme, thereby improving prediction accuracy.
To accurately identify catalytic domains, molecular docking methods are typically used to simulate interactions between enzymes and substrates and to obtain the structure of enzyme-substrate complexes [9–10]. Based on this principle, this study proposes a deep learning method for structure-based prediction of enzyme Km values. The model takes the structural files of enzymes and substrates as input. It can either utilize the entire enzyme structure in combination with substrate features to predict Km values, or focus specifically on the catalytic domain and substrate features for prediction.
2. Methods
2.1. Data acquisition
To predict enzyme Km values, it is necessary to collect data related to Km values as well as the three-dimensional structures of enzymes and substrates. In this study, relevant information was carefully selected and integrated from multiple authoritative databases. The specific steps are as follows:
1) Comprehensive Retrieval of Enzyme Information: Using the Python programming language [11], enzyme-related data were retrieved from the BRENDA [12–13] and SABIO-RK [14] databases, including Km values, substrate names, source organisms, and UniProt IDs.
2) Filtering for Wild-Type Enzymes [15]: Among the collected enzyme entries, only those describing wild-type enzymes were retained. Wild-type enzymes, having undergone long-term natural selection and optimization, exhibit relatively stable and reliable structures and functions, making them more suitable for the purposes of this study.
3) Accurate Matching of Substrate Information: Substrate names were used to retrieve their corresponding Simplified Molecular Input Line Entry System (SMILES) representations.
4) Standardization of Km Values: To facilitate subsequent data processing and analysis, all Km values were transformed using the base-10 logarithm (log₁₀), thereby normalizing them to a consistent numerical range. This transformation also helps mitigate the influence of extreme and outlier values on the analysis.
5) Acquisition of Structural Files: Structural files for enzymes and substrates were obtained from the UniProt [16] and PubChem [17] databases. Entries without corresponding structural data were excluded from the dataset.
2.2. Data processing via molecular docking
Features within the catalytic domain have a significant impact on enzyme Km values. To perform Km prediction based on these features, it is first necessary to obtain the enzyme-substrate complex structure. Molecular docking focuses on studying the interactions between receptors and ligands. By simulating the binding process between enzymes and substrates, it is possible to obtain their complex structures. Currently, there are various molecular docking methods, each employing different algorithms and yielding varying results. Choosing an appropriate docking method is crucial for accurately modeling enzyme-substrate interactions and subsequently identifying the catalytic domain.
Two key components in molecular docking are sampling methods and scoring functions. Sampling methods explore different orientations and conformations of ligands within the receptor’s binding site, aiming to find the optimal binding pose to maximize interactions. Scoring functions evaluate the quality of each binding pose by assigning a numerical score. Wang et al [18]. systematically evaluated the performance of 10 docking approaches—5 commercial tools (LigandFit, Glide, GOLD, MOE Dock, and Surflex-Dock) and 5 academic tools (AutoDock, AutoDock Vina, LeDock, rDock, and UCSF Dock)—by assessing the accuracy of their sampling methods and scoring functions. Their results showed that GOLD and LeDock offered the best sampling performance, while AutoDock Vina provided the most accurate scoring capability. Therefore, this study selected AutoDock Vina [19] and LeDock [20], two top-performing academic software tools in both sampling and scoring, for molecular docking analysis.
2.3. Feature representation of data
The input to the proposed model consists of structural files of enzymes and substrates, which contain their respective three-dimensional (3D) structural information. By parsing these structural files, the 3D data can be represented in the form of graphs, which serve as inputs to the deep learning model.
For proteins, their 3D structures were parsed using a Python-based program. Specifically, the program iterated through the amino acids in each protein to extract key information such as sequence data, atomic coordinates, and torsion angles between atoms. This process enabled the representation of a protein as a 3D graph [21] G=(V,E), where each node v∈V corresponds to an amino acid residue and carries both scalar and vector features. Additionally, spatial distances were computed between each amino acid residue and all others. For each residue, the 30 closest neighboring residues were identified, and edges were created between them to form the edge set E. Each edge e∈E also contains scalar and vector features.
Given that the catalytic domain plays a crucial role in enzyme-substrate binding but is also influenced by the global structure of the enzyme, two strategies were employed when converting the protein’s 3D structure into graph representations: In the first method, all amino acids in the enzyme structure are traversed, and the entire protein is represented as a graph (Figure 1(a)). This approach considers the global structure and properties of the enzyme, thereby offering more comprehensive information for prediction. The second method focuses exclusively on amino acids located within the specific structural domain involved in enzyme-substrate interactions or catalysis [22] (Figure 1(b)). Only the amino acids within the defined circular region are considered and converted into a graph. This localized representation more precisely reflects the regions of the enzyme responsible for substrate binding and catalytic activity.
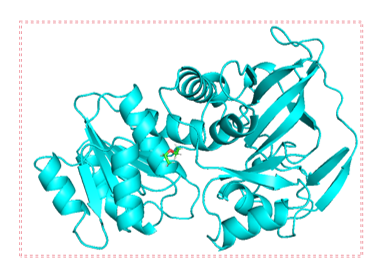
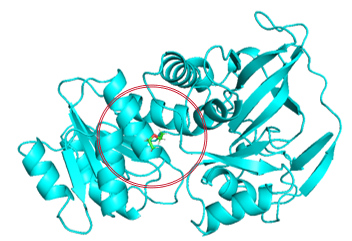
Figure 1: Traversal of amino acid regions
For substrate molecules, the RDKit toolkit [23] was used to parse their structural files. Chemical features of the nodes (atoms) and edges (bonds) were extracted using TorchDrug [24], allowing each substrate to be represented as a graph G=(V,E), where each node v∈V represents an atom and each edge e∈E denotes a bond within the molecule.
2.4. Model architecture overview
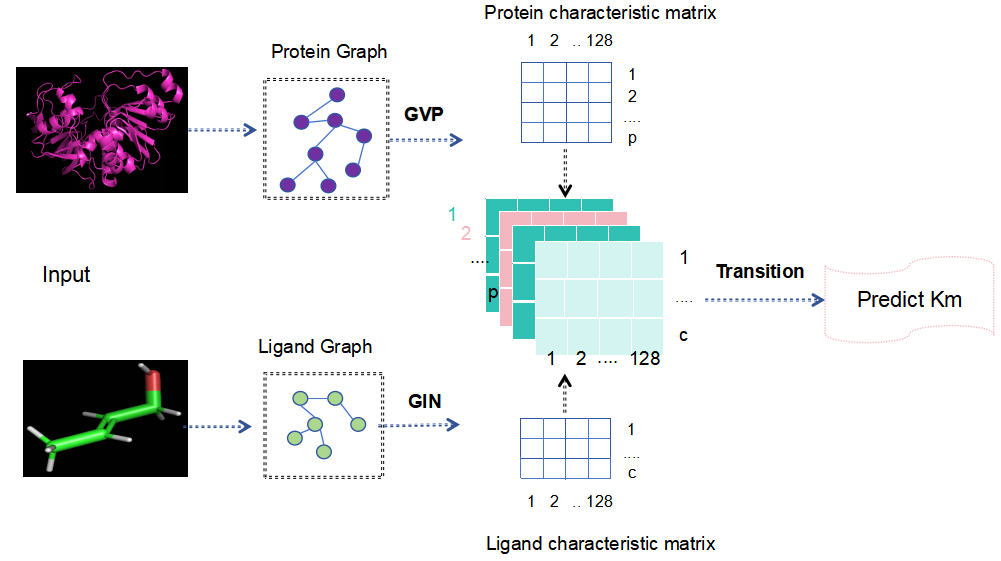
The overall architecture of the model is shown in Figure 2. The model is designed to predict enzyme Km values based on the structural files of enzymes and substrates. First, the structural files are converted into graph representations using Python, enabling subsequent feature extraction and learning. The protein graph is then processed by a Geometric Vector Perceptron (GVP), which focuses on learning key features from the enzyme. Meanwhile, the substrate graph is processed by a Graph Isomorphism Network (GIN), which captures structural features of the substrate. Once the features of both enzyme and substrate are obtained, they are fused and passed into a Transition Network for training and prediction.
The GVP, proposed by Jing et al [25]., is a network architecture based on a message-passing mechanism and is specifically designed for tasks involving 3D data and shape recognition. Since a protein’s three-dimensional structure is essential to its biological function, the GVP is particularly adept at utilizing 3D coordinate information to accurately capture structural features. It has shown excellent performance in the field of protein design. In this study, the GVP is employed to extract features from proteins and outputs a feature matrix of shape (p,128), where p denotes the number of amino acid residues.
The GIN, developed by Xu et al [26]., is a novel graph neural network architecture consisting of multiple graph convolution layers. It incorporates graph isomorphism into the network structure and utilizes a learnable node aggregation and update mechanism to extract global information from local features. The core innovation of GIN lies in its node feature update strategy, which combines the features of a node with those of its neighbors in the previous layer and applies a multilayer perceptron (MLP) for spatial transformation and feature updating. This mechanism retains local information while integrating broader contextual data. The update rule is defined as follows:
In Equation (1), ϵ is a learnable parameter, and
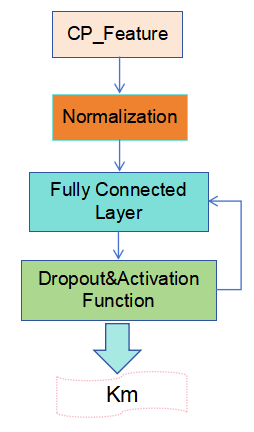
A Transition Network was constructed using multiple techniques to optimize the model’s performance and enhance its stability, as illustrated in Figure 3. First, the enzyme and substrate features are combined to form a compound feature matrix of shape (p,c,128), ensuring that each amino acid feature in the enzyme is associated with every atomic feature in the substrate. Next, the input layer is normalized to improve data stability. A fully connected (dense) layer is then introduced as the hidden layer, with both the input and output dimensions set to 128. This layer incorporates a Dropout function and an activation function to reduce model complexity and enhance nonlinearity, helping the model learn and identify patterns more effectively. Finally, a fully connected output layer with an output dimension of 1 is defined. This layer receives and integrates the features from the hidden layer and is responsible for predicting the enzyme’s Km value, thereby completing the end-to-end transformation from input to output.
2.5. Evaluation metrics
This study adopts several evaluation metrics to assess the performance of the model, including the coefficient of determination (R²), the Pearson correlation coefficient (Pearson), and the root mean square error (RMSE).
R² measures the proportion of variance in the observed data that is explained by the model. Its value ranges from 0 to 1. An R² of 1 indicates that the model perfectly explains the variance in the data, whereas an R² of 0 indicates that the model fails to explain any of the variance. The calculation is shown in Equation (2):
Where:
The Pearson correlation coefficient measures the strength and direction of the linear relationship between two variables. Its value ranges from -1 to 1. A value of 1 indicates a perfect positive linear correlation, -1 indicates a perfect negative linear correlation, and 0 indicates no linear correlation. The formula is given in Equation (3):
Pearson=
RMSE quantifies the average magnitude of the prediction error, serving as an indicator of model accuracy. It is computed as the square root of the mean of the squared differences between predicted and actual values. A smaller RMSE indicates better predictive accuracy. The calculation is shown in Equation (4):
RMSE =
3. Results and discussion
In this study, the trained model was first evaluated on the BRENDA test set to assess its ability to predict enzyme Km values from structural information. Additionally, the two proposed strategies for selecting protein feature regions were compared and analyzed. Further evaluation was conducted using an independently collected test set from the SABIO-RK database. Finally, a comparison was made with the method proposed by Kroll et al.
3.1. Results of experimental data collection
After processing all data entries for wild-type enzymes in the BRENDA database, a dataset containing 11,112 entries was successfully constructed. To ensure fairness in model training and evaluation, the dataset was randomly divided into a training set and a test set. Specifically, 9,179 entries were used for training and 1,933 entries were used for testing. This dataset, derived from BRENDA, served as the primary data source for training and evaluating the model.
To further verify the model’s predictive capability, an independent dataset containing 447 entries was obtained from the SABIO-RK database. This dataset is completely independent of the BRENDA data and includes only entries that are not present in BRENDA. It was used as an external test set and served as the basis for comparison with the method developed by Kroll et al.
3.2. Prediction results and discussion on the BRENDA test set
Using the first protein traversal strategy described in Section 2.3, the model was trained and evaluated, with results shown in Model 1 of Table 1. For the second traversal strategy, it was necessary to first obtain enzyme-substrate complex structures via molecular docking. In this study, AutoDock Vina and LeDock were adopted as the docking tools. Based on the docking results, two different local catalytic domain sizes were tested: A local region with a 1.5 nm radius centered around the substrate's docking position. A larger region with a 2.0 nm radius. The performance of the models trained with these local regions is presented as Models 2, 3, 4, and 5 in Table 1.
|
Model |
Feature Region Selection Method |
Docking Method |
Region Radius/nm |
Pearson |
RMSE |
R2 |
|
Model 1 |
Full Structure |
0.706 |
0.873 |
0.479 |
||
|
Model 2 |
Local Structure |
Vina |
1.5 |
0.704 |
0.877 |
0.474 |
|
Model 3 |
Local Structure |
LeDock |
1.5 |
0.672 |
0.901 |
0.445 |
|
Model 4 |
Local Structure |
Vina |
2 |
0.717 |
0.854 |
0.501 |
|
Model 5 |
Local Structure |
LeDock |
2 |
0.691 |
0.883 |
0.467 |
When selecting the radius for the local region, it is important to note that proteins typically contain multiple domains, each comprising approximately 100 to 200 amino acids. These domains can independently fold and interact with ligands. A spherical region with a radius of 1.5 nm generally includes around 100 amino acids, while expanding the radius to 2.0 nm usually encompasses about 200 amino acids.
As shown in Table 1, Model 4 achieved the best performance, followed by Model 1. This indicates that using local enzyme region features for training yields better prediction results than using the entire enzyme structure. The inferior performance of the full-structure approach may be attributed to the inclusion of numerous irrelevant features unrelated to the Km value, making it difficult for the model to distinguish between essential and redundant information when training data is limited. In contrast, focusing on the structural domain involved in enzyme-substrate interactions helps eliminate extraneous features, leading to more accurate predictions on the test set.
At the same time, Table 1 also shows that while models trained on local region features perform best overall, the results are affected by the size of the selected region and the molecular docking method used. Specifically, when the radius of the selected region is 2.0 nm, model performance is better than with a 1.5 nm radius. Therefore, this approach requires multiple experiments to determine the optimal region size.
Under otherwise identical conditions, models using AutoDock Vina for molecular docking produced significantly better results than those using LeDock. This advantage stems primarily from AutoDock Vina’s superior scoring function. Given that this study involves batch docking tasks, the substrates with the highest docking scores from each software were selected for analysis. AutoDock Vina's scoring function offers more accurate evaluations of docking quality. Although LeDock exhibits excellent sampling capabilities, its top-scoring results do not always correspond to the best actual binding conformations. Therefore, AutoDock Vina is more suitable for large-scale docking tasks. This finding underscores the importance of selecting an appropriate molecular docking tool to enhance prediction accuracy.
3.3. Evaluation of model performance on the independent SABIO-RK test set
The model developed in this study was trained and tested using data from the BRENDA database. To assess the model's predictive performance and generalization capability, it is essential to test it on data from alternative sources. Therefore, an independent test set was constructed using data from the SABIO-RK database. The prediction results of the model on this dataset are presented in Table 2.
|
Model |
Feature Region Selection Method |
Docking Method |
Region Radius/nm |
Pearson |
RMSE |
R2 |
|
Model 1 |
Full Structure |
0.637 |
0.992 |
0.391 |
||
|
Model 2 |
Local Structure |
Vina |
1.5 |
0.594 |
1.034 |
0.339 |
|
Model 3 |
Local Structure |
LeDock |
1.5 |
0.628 |
1.009 |
0.370 |
|
Model 4 |
Local Structure |
Vina |
2 |
0.675 |
0.94 |
0.453 |
|
Model 5 |
Local Structure |
LeDock |
2 |
0.63 |
0.993 |
0.390 |
As shown in Table 2, the models trained on the BRENDA dataset all achieved satisfactory predictive performance on the SABIO-RK test set, fully demonstrating the stability and accuracy of the proposed model. Among them, Model 4 again achieved the best performance on the SABIO-RK dataset, significantly outperforming all other models. On the other hand, Model 2 performed the worst, which highlights the need for caution when evaluating model performance—even if a model performs well on the BRENDA test set, its generalizability must be validated with additional data and experiments to confirm its effectiveness and stability.
3.4. Comparison of model performance
To further validate the model’s performance, we compared it with the model developed by Kroll et al., which was also trained on data from the BRENDA database and evaluated using the SABIO-RK test set. The reported evaluation metrics of their model on the SABIO-RK dataset are as follows: Pearson correlation coefficient: 0.5905; RMSE: 1.0508; R²: 0.317.
By contrast, the model proposed in this study demonstrated significantly better performance on the same test set. Specifically, Model 4 achieved an R² value of 0.453, which is markedly higher than the R² of 0.317 reported by Kroll et al. This substantial difference underscores the superior predictive accuracy of the model developed in this work.
4. Conclusion
This study proposes a novel approach for predicting enzyme Michaelis constant (Km) values based on structural information, and successfully verifies its feasibility and effectiveness. Compared with sequence-based prediction methods, the proposed structure-based approach offers several distinctive advantages:
1) Closer correlation between structure and function: The three-dimensional structures of enzymes and substrates directly reflect their functional properties, providing a more precise foundation for predicting Km values. Given reliable structural data, this approach enables more accurate predictions.
2) Focusing on catalytic domains enhances prediction accuracy: By analyzing only the catalytic domain of the enzyme and its interaction with the substrate, the model avoids interference from non-essential structural features. This significantly improves prediction precision—something that sequence-based models struggle to achieve—highlighting the unique strength of structure-based prediction.
3) Stronger generalization capability: Experimental results show that the proposed model achieved an R² of 0.453 on the independent SABIO-RK dataset, substantially outperforming Kroll et al.’s sequence-based method (R² = 0.317). This demonstrates the model’s strong generalizability, making it suitable for diverse datasets from different sources and with varying characteristics, thus offering greater robustness and reliability.
To further improve the accuracy of Km value predictions, future research could focus on the following directions: Expanding and refining datasets: More comprehensive and higher-quality data is essential for effective model training, which in turn leads to enhanced prediction accuracy. Developing more effective feature selection strategies: Precisely identifying the core features most relevant to Km, while eliminating irrelevant or redundant information, will further boost both prediction accuracy and the model’s generalization performance.
References
[1]. Borger, S., Liebermeister, W., & Klipp, E. (2006). Prediction of enzyme kinetic parameters based on statistical learning. Genome Informatics, 17(1), 80–87. https: //doi.org/10.11234/gi1990.17.80
[2]. Heckmann, D., Lloyd, C. J., Mih, N., Ha, Y., Zielinski, D. C., Haiman, Z. B., Desouki, A., Lercher, M. J., & Palsson, B. O. (2018). Machine learning applied to enzyme turnover numbers reveals protein structural correlates and improves metabolic models. Nature Communications, 9(1), 5252. https: //doi.org/10.1038/s41467-018-07652-6
[3]. Kroll, A., Engqvist, M. K. M., Lercher, M. J., & Heckmann, D. (2021). Deep learning allows genome-scale prediction of Michaelis constants from structural features. PLoS Biology, 19(10), e3001402. https: //doi.org/10.1371/journal.pbio.3001402
[4]. Alley, E. C., Khimulya, G., Biswas, S., AlQuraishi, M., & Church, G. M. (2019). Unified rational protein engineering with sequence-based deep representation learning. Nature Methods, 16(12), 1315–1322. https: //doi.org/10.1038/s41592-019-0598-1
[5]. Chen, T., & Guestrin, C. (2016). XGBoost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (pp. 785–794). https: //doi.org/10.1145/2939672.2939785
[6]. Yu, H., Deng, H., He, J., Keasling, J. D., & Luo, X. (2023). UniKP: A unified framework for the prediction of enzyme kinetic parameters. Nature Communications, 14(1), 8211. https: //doi.org/10.1038/s41467-023-44113-1
[7]. Elnaggar, A., Heinzinger, M., Dallago, C., Rehawi, G., Wang, Y., Jones, L., ... & Rost, B. (2021). ProtTrans: Towards cracking the language of life’s code through self-supervised learning. IEEE Transactions on Pattern Analysis and Machine Intelligence, 44, 7112–7127.
[8]. Honda, S., Shi, S., & Ueda, H. R. (2019). SMILES transformer: Pre-trained molecular fingerprint for low-data drug discovery. arXiv Preprint arXiv: 1911.04738. https: //doi.org/10.48550/arXiv.1911.04738
[9]. Jiménez, J., Skalic, M., Martinez-Rosell, G., & De Fabritiis, G. (2018). KDEEP: Protein–ligand absolute binding affinity prediction via 3D-convolutional neural networks. Journal of Chemical Information and Modeling, 58(2), 287–296. https: //doi.org/10.1021/acs.jcim.7b00650
[10]. Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. https: //doi.org/10.1002/jcc.21256
[11]. Van Rossum, G., & Drake, F. L. (1995). Python reference manual. Centrum voor Wiskunde en Informatica.
[12]. Schomburg, I., Chang, A., Placzek, S., Söhngen, C., Rother, M., Lang, M., ... & Schomburg, D. (2012). BRENDA in 2013: Integrated reactions, kinetic data, enzyme function data, improved disease classification: New options and contents in BRENDA. Nucleic Acids Research, 41(D1), D764–D772. https: //doi.org/10.1093/nar/gks1049
[13]. Jeske, L., Placzek, S., Schomburg, I., Chang, A., & Schomburg, D. (2019). BRENDA in 2019: A European ELIXIR core data resource. Nucleic Acids Research, 47(D1), D542–D549. https: //doi.org/10.1093/nar/gky1048
[14]. Wittig, U., Rey, M., Weidemann, A., Kania, R., & Müller, W. (2018). SABIO-RK: An updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Research, 46(D1), D656–D660. https: //doi.org/10.1093/nar/gkx1065
[15]. Bar-Even, A., Noor, E., Savir, Y., Liebermeister, W., Davidi, D., Tawfik, D. S., & Milo, R. (2011). The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry, 50(21), 4402–4410. https: //doi.org/10.1021/bi2002289
[16]. UniProt Consortium. (2023). UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Research, 51(D1), 523–531. https: //doi.org/10.1093/nar/gkac1052
[17]. Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., ... & Bolton, E. E. (2019). PubChem 2019 update: Improved access to chemical data. Nucleic Acids Research, 47(D1), D1102–D1109. https: //doi.org/10.1093/nar/gky1033
[18]. Wang, Z., Sun, H., Yao, X., Li, D., Xu, L., Li, Y., ... & Hou, T. (2016). Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Physical Chemistry Chemical Physics, 18(18), 12964–12975. https: //doi.org/10.1039/C6CP01555G
[19]. Eberhardt, J., Santos-Martins, D., Tillack, A. F., & Forli, S. (2021). AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. Journal of Chemical Information and Modeling, 61(8), 3891–3898. https: //doi.org/10.1021/acs.jcim.1c00203
[20]. Zhao, H., & Huang, D. (2011). Hydrogen bonding penalty upon ligand binding. PLoS ONE, 6(6), e19923. https: //doi.org/10.1371/journal.pone.0019923
[21]. Jing, B., Eismann, S., Soni, P. N., & Dror, R. O. (2021). Equivariant graph neural networks for 3D macromolecular structure. arXiv Preprint arXiv: 2106.03843. https: //doi.org/10.48550/arXiv.2106.03843
[22]. Furnham, N., Holliday, G. L., De Beer, T. A., Jacobsen, J. O., Pearson, W. R., & Thornton, J. M. (2014). The Catalytic Site Atlas 2.0: Cataloging catalytic sites and residues identified in enzymes. Nucleic Acids Research, 42(D1), D485–D489. https: //doi.org/10.1093/nar/gkt1243
[23]. Landrum, G. (2019). RDKit: Open-source cheminformatics from machine learning to chemical registration. Abstracts of Papers of the American Chemical Society.
[24]. Zhu, Z., Shi, C., Zhang, Z., Liu, S., Xu, M., Yuan, X., ... & Tang, J. (2022). TorchDrug: A powerful and flexible machine learning platform for drug discovery. arXiv Preprint arXiv: 2202.08320. https: //doi.org/10.48550/arXiv.2202.08320
[25]. Jing, B., Eismann, S., Suriana, P., Townshend, R. J. L., & Dror, R. (2009). Learning from protein structure with geometric vector perceptrons. arXiv Preprint arXiv: 2009.01411. https: //doi.org/10.48550/arXiv.2009.01411
[26]. Xu, K., Hu, W., Leskovec, J., & Jegelka, S. (2018). How powerful are graph neural networks? arXiv Preprint arXiv: 1810.00826. https: //doi.org/10.48550/arXiv.1810.00826
Cite this article
Xie,J.;Yan,M. (2025). Prediction of Enzyme Michaelis Constant (Km) Based on Deep Learning. Theoretical and Natural Science,121,7-16.
Data availability
The datasets used and/or analyzed during the current study will be available from the authors upon reasonable request.
Disclaimer/Publisher's Note
The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of EWA Publishing and/or the editor(s). EWA Publishing and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
About volume
Volume title: Proceedings of the 5th International Conference on Biological Engineering and Medical Science
© 2024 by the author(s). Licensee EWA Publishing, Oxford, UK. This article is an open access article distributed under the terms and
conditions of the Creative Commons Attribution (CC BY) license. Authors who
publish this series agree to the following terms:
1. Authors retain copyright and grant the series right of first publication with the work simultaneously licensed under a Creative Commons
Attribution License that allows others to share the work with an acknowledgment of the work's authorship and initial publication in this
series.
2. Authors are able to enter into separate, additional contractual arrangements for the non-exclusive distribution of the series's published
version of the work (e.g., post it to an institutional repository or publish it in a book), with an acknowledgment of its initial
publication in this series.
3. Authors are permitted and encouraged to post their work online (e.g., in institutional repositories or on their website) prior to and
during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work (See
Open access policy for details).
References
[1]. Borger, S., Liebermeister, W., & Klipp, E. (2006). Prediction of enzyme kinetic parameters based on statistical learning. Genome Informatics, 17(1), 80–87. https: //doi.org/10.11234/gi1990.17.80
[2]. Heckmann, D., Lloyd, C. J., Mih, N., Ha, Y., Zielinski, D. C., Haiman, Z. B., Desouki, A., Lercher, M. J., & Palsson, B. O. (2018). Machine learning applied to enzyme turnover numbers reveals protein structural correlates and improves metabolic models. Nature Communications, 9(1), 5252. https: //doi.org/10.1038/s41467-018-07652-6
[3]. Kroll, A., Engqvist, M. K. M., Lercher, M. J., & Heckmann, D. (2021). Deep learning allows genome-scale prediction of Michaelis constants from structural features. PLoS Biology, 19(10), e3001402. https: //doi.org/10.1371/journal.pbio.3001402
[4]. Alley, E. C., Khimulya, G., Biswas, S., AlQuraishi, M., & Church, G. M. (2019). Unified rational protein engineering with sequence-based deep representation learning. Nature Methods, 16(12), 1315–1322. https: //doi.org/10.1038/s41592-019-0598-1
[5]. Chen, T., & Guestrin, C. (2016). XGBoost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (pp. 785–794). https: //doi.org/10.1145/2939672.2939785
[6]. Yu, H., Deng, H., He, J., Keasling, J. D., & Luo, X. (2023). UniKP: A unified framework for the prediction of enzyme kinetic parameters. Nature Communications, 14(1), 8211. https: //doi.org/10.1038/s41467-023-44113-1
[7]. Elnaggar, A., Heinzinger, M., Dallago, C., Rehawi, G., Wang, Y., Jones, L., ... & Rost, B. (2021). ProtTrans: Towards cracking the language of life’s code through self-supervised learning. IEEE Transactions on Pattern Analysis and Machine Intelligence, 44, 7112–7127.
[8]. Honda, S., Shi, S., & Ueda, H. R. (2019). SMILES transformer: Pre-trained molecular fingerprint for low-data drug discovery. arXiv Preprint arXiv: 1911.04738. https: //doi.org/10.48550/arXiv.1911.04738
[9]. Jiménez, J., Skalic, M., Martinez-Rosell, G., & De Fabritiis, G. (2018). KDEEP: Protein–ligand absolute binding affinity prediction via 3D-convolutional neural networks. Journal of Chemical Information and Modeling, 58(2), 287–296. https: //doi.org/10.1021/acs.jcim.7b00650
[10]. Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. https: //doi.org/10.1002/jcc.21256
[11]. Van Rossum, G., & Drake, F. L. (1995). Python reference manual. Centrum voor Wiskunde en Informatica.
[12]. Schomburg, I., Chang, A., Placzek, S., Söhngen, C., Rother, M., Lang, M., ... & Schomburg, D. (2012). BRENDA in 2013: Integrated reactions, kinetic data, enzyme function data, improved disease classification: New options and contents in BRENDA. Nucleic Acids Research, 41(D1), D764–D772. https: //doi.org/10.1093/nar/gks1049
[13]. Jeske, L., Placzek, S., Schomburg, I., Chang, A., & Schomburg, D. (2019). BRENDA in 2019: A European ELIXIR core data resource. Nucleic Acids Research, 47(D1), D542–D549. https: //doi.org/10.1093/nar/gky1048
[14]. Wittig, U., Rey, M., Weidemann, A., Kania, R., & Müller, W. (2018). SABIO-RK: An updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Research, 46(D1), D656–D660. https: //doi.org/10.1093/nar/gkx1065
[15]. Bar-Even, A., Noor, E., Savir, Y., Liebermeister, W., Davidi, D., Tawfik, D. S., & Milo, R. (2011). The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry, 50(21), 4402–4410. https: //doi.org/10.1021/bi2002289
[16]. UniProt Consortium. (2023). UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Research, 51(D1), 523–531. https: //doi.org/10.1093/nar/gkac1052
[17]. Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., ... & Bolton, E. E. (2019). PubChem 2019 update: Improved access to chemical data. Nucleic Acids Research, 47(D1), D1102–D1109. https: //doi.org/10.1093/nar/gky1033
[18]. Wang, Z., Sun, H., Yao, X., Li, D., Xu, L., Li, Y., ... & Hou, T. (2016). Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Physical Chemistry Chemical Physics, 18(18), 12964–12975. https: //doi.org/10.1039/C6CP01555G
[19]. Eberhardt, J., Santos-Martins, D., Tillack, A. F., & Forli, S. (2021). AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. Journal of Chemical Information and Modeling, 61(8), 3891–3898. https: //doi.org/10.1021/acs.jcim.1c00203
[20]. Zhao, H., & Huang, D. (2011). Hydrogen bonding penalty upon ligand binding. PLoS ONE, 6(6), e19923. https: //doi.org/10.1371/journal.pone.0019923
[21]. Jing, B., Eismann, S., Soni, P. N., & Dror, R. O. (2021). Equivariant graph neural networks for 3D macromolecular structure. arXiv Preprint arXiv: 2106.03843. https: //doi.org/10.48550/arXiv.2106.03843
[22]. Furnham, N., Holliday, G. L., De Beer, T. A., Jacobsen, J. O., Pearson, W. R., & Thornton, J. M. (2014). The Catalytic Site Atlas 2.0: Cataloging catalytic sites and residues identified in enzymes. Nucleic Acids Research, 42(D1), D485–D489. https: //doi.org/10.1093/nar/gkt1243
[23]. Landrum, G. (2019). RDKit: Open-source cheminformatics from machine learning to chemical registration. Abstracts of Papers of the American Chemical Society.
[24]. Zhu, Z., Shi, C., Zhang, Z., Liu, S., Xu, M., Yuan, X., ... & Tang, J. (2022). TorchDrug: A powerful and flexible machine learning platform for drug discovery. arXiv Preprint arXiv: 2202.08320. https: //doi.org/10.48550/arXiv.2202.08320
[25]. Jing, B., Eismann, S., Suriana, P., Townshend, R. J. L., & Dror, R. (2009). Learning from protein structure with geometric vector perceptrons. arXiv Preprint arXiv: 2009.01411. https: //doi.org/10.48550/arXiv.2009.01411
[26]. Xu, K., Hu, W., Leskovec, J., & Jegelka, S. (2018). How powerful are graph neural networks? arXiv Preprint arXiv: 1810.00826. https: //doi.org/10.48550/arXiv.1810.00826